

WNT Signaling Perturbations Underlie the Genetic Heterogeneity of Robinow Syndrome
- PMID: 29276006
- PMCID: PMC5777383
- DOI: 10.1016/j.ajhg.2017.10.002
WNT Signaling Perturbations Underlie the Genetic Heterogeneity of Robinow Syndrome
Abstract
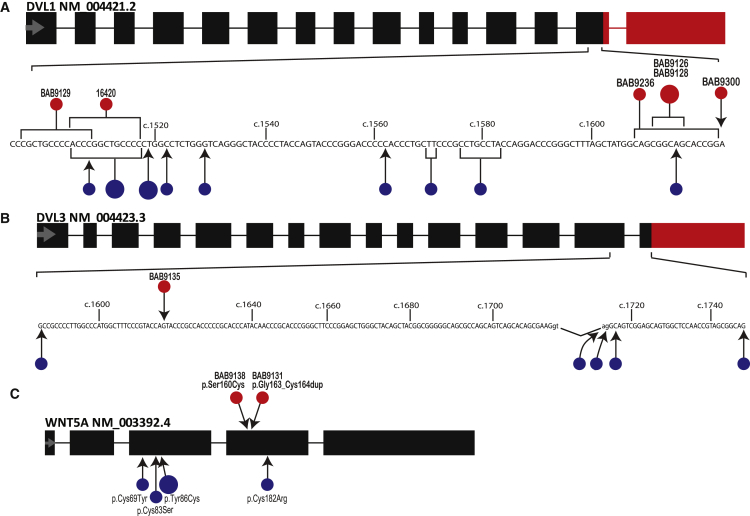
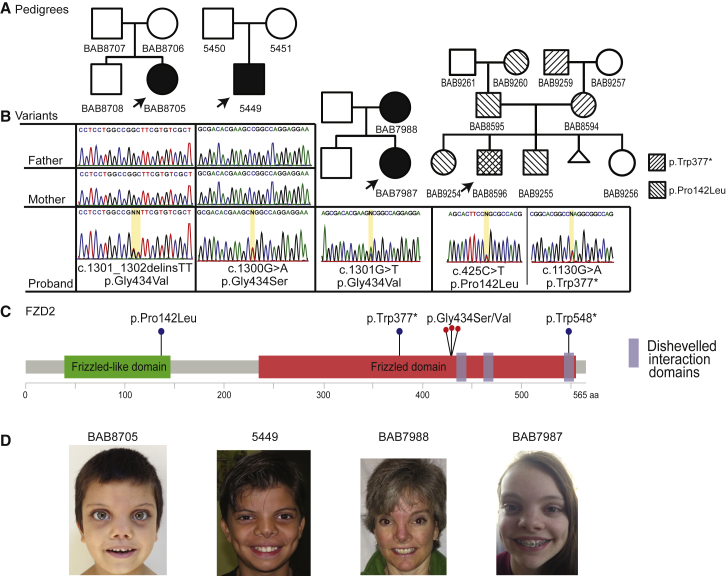
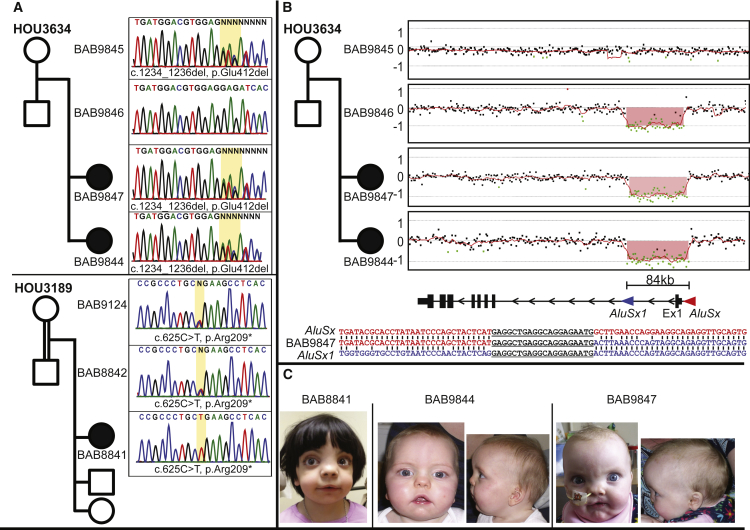
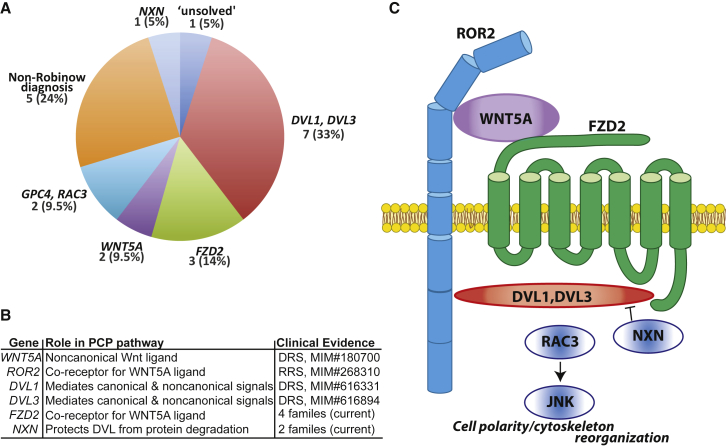
Locus heterogeneity characterizes a variety of skeletal dysplasias often due to interacting or overlapping signaling pathways. Robinow syndrome is a skeletal disorder historically refractory to molecular diagnosis, potentially stemming from substantial genetic heterogeneity. All current known pathogenic variants reside in genes within the noncanonical Wnt signaling pathway including ROR2, WNT5A, and more recently, DVL1 and DVL3. However, ∼70% of autosomal-dominant Robinow syndrome cases remain molecularly unsolved. To investigate this missing heritability, we recruited 21 families with at least one family member clinically diagnosed with Robinow or Robinow-like phenotypes and performed genetic and genomic studies. In total, four families with variants in FZD2 were identified as well as three individuals from two families with biallelic variants in NXN that co-segregate with the phenotype. Importantly, both FZD2 and NXN are relevant protein partners in the WNT5A interactome, supporting their role in skeletal development. In addition to confirming that clustered -1 frameshifting variants in DVL1 and DVL3 are the main contributors to dominant Robinow syndrome, we also found likely pathogenic variants in candidate genes GPC4 and RAC3, both linked to the Wnt signaling pathway. These data support an initial hypothesis that Robinow syndrome results from perturbation of the Wnt/PCP pathway, suggest specific relevant domains of the proteins involved, and reveal key contributors in this signaling cascade during human embryonic development. Contrary to the view that non-allelic genetic heterogeneity hampers gene discovery, this study demonstrates the utility of rare disease genomic studies to parse gene function in human developmental pathways.
Keywords: Frizzled; dual molecular diagnosis; human embryonic development; skeletal dysplasia.
Copyright © 2017 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Robinow M., Silverman F.N., Smith H.D. A newly recognized dwarfing syndrome. Am. J. Dis. Child. 1969;117:645–651. - PubMed
-
- van Bokhoven H., Celli J., Kayserili H., van Beusekom E., Balci S., Brussel W., Skovby F., Kerr B., Percin E.F., Akarsu N., Brunner H.G. Mutation of the gene encoding the ROR2 tyrosine kinase causes autosomal recessive Robinow syndrome. Nat. Genet. 2000;25:423–426. - PubMed
-
- Afzal A.R., Rajab A., Fenske C.D., Oldridge M., Elanko N., Ternes-Pereira E., Tüysüz B., Murday V.A., Patton M.A., Wilkie A.O., Jeffery S. Recessive Robinow syndrome, allelic to dominant brachydactyly type B, is caused by mutation of ROR2. Nat. Genet. 2000;25:419–422. - PubMed
-
- Oishi I., Suzuki H., Onishi N., Takada R., Kani S., Ohkawara B., Koshida I., Suzuki K., Yamada G., Schwabe G.C. The receptor tyrosine kinase Ror2 is involved in non-canonical Wnt5a/JNK signalling pathway. Genes Cells. 2003;8:645–654. - PubMed
Publication types
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
