

BP180 Is Critical in the Autoimmunity of Bullous Pemphigoid
- PMID: 29276517
- PMCID: PMC5727044
- DOI: 10.3389/fimmu.2017.01752
BP180 Is Critical in the Autoimmunity of Bullous Pemphigoid
Abstract
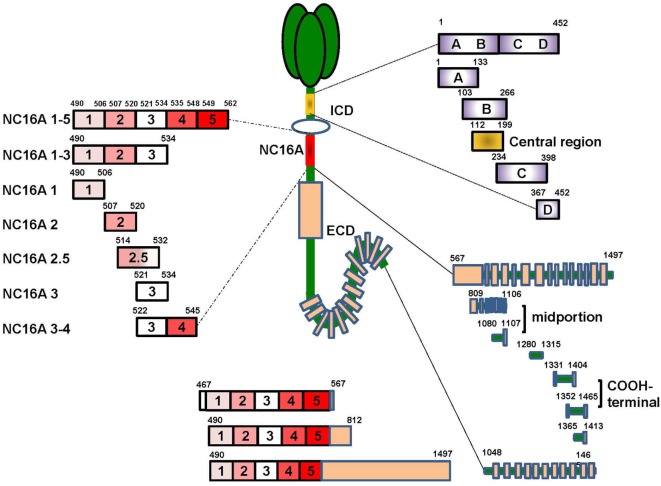
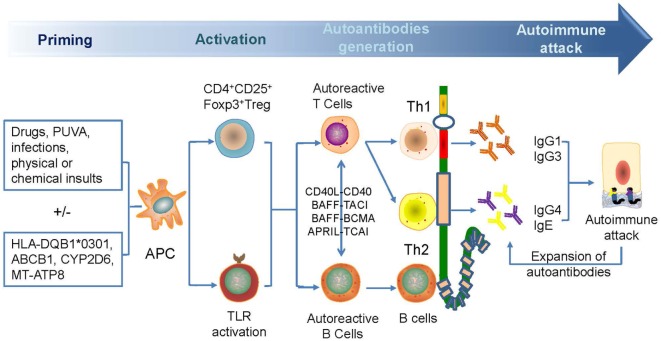
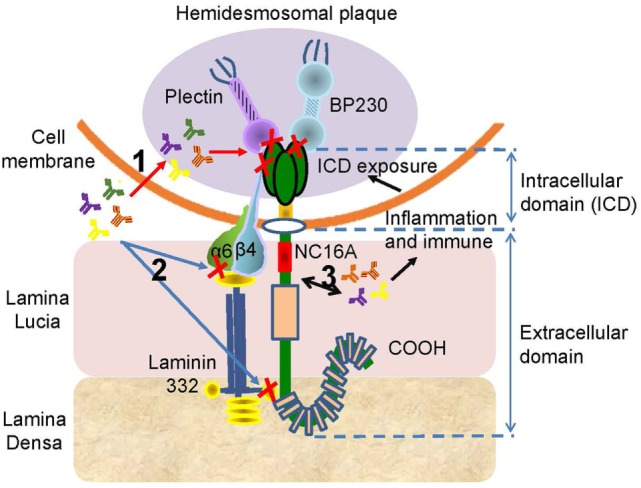
Bullous pemphigoid (BP) is by far the most common autoimmune blistering dermatosis that mainly occurs in the elderly. The BP180 is a transmembrane glycoprotein, which is highly immunodominant in BP. The structure and location of BP180 indicate that it is a significant autoantigen and plays a key role in blister formation. Autoantibodies from BP patients react with BP180, which leads to its degradation and this has been regarded as the central event in BP pathogenesis. The consequent blister formation involves the activation of complement-dependent or -independent signals, as well as inflammatory pathways induced by BP180/anti-BP180 autoantibody interaction. As a multi-epitope molecule, BP180 can cause dermal-epidermal separation via combining each epitope with specific immunoglobulin, which also facilitates blister formation. In addition, some inflammatory factors can directly deplete BP180, thereby leading to fragility of the dermal-epidermal junction and blister formation. This review summarizes recent investigations on the role of BP180 in BP pathogenesis to determine the potential targets for the treatment of patients with BP.
Keywords: BP180; autoantibody; bullous pemphigoid; cytokine; dermal–epidermal junction.
Figures


Similar articles
-
Update on the pathogenesis of bullous pemphigoid: an autoantibody-mediated blistering disease targeting collagen XVII.J Dermatol Sci. 2014 Mar;73(3):179-86. doi: 10.1016/j.jdermsci.2013.12.001. Epub 2013 Dec 27. J Dermatol Sci. 2014. PMID: 24434029 Review.
-
Evidence for a role of eosinophils in blister formation in bullous pemphigoid.Allergy. 2017 Jul;72(7):1105-1113. doi: 10.1111/all.13131. Epub 2017 Mar 1. Allergy. 2017. PMID: 28135772
-
Levels of anti-BP180 NC16A IgE do not correlate with severity of disease in the early stages of bullous pemphigoid.Arch Dermatol Res. 2015 Nov;307(9):849-54. doi: 10.1007/s00403-015-1598-3. Epub 2015 Sep 24. Arch Dermatol Res. 2015. PMID: 26404084
-
Respective contribution of neutrophil elastase and matrix metalloproteinase 9 in the degradation of BP180 (type XVII collagen) in human bullous pemphigoid.J Invest Dermatol. 2001 Nov;117(5):1091-6. doi: 10.1046/j.0022-202x.2001.01521.x. J Invest Dermatol. 2001. PMID: 11710917
-
An animal model of bullous pemphigoid: what can it teach us?Proc Assoc Am Physicians. 1995 Jul;107(2):237-41. Proc Assoc Am Physicians. 1995. PMID: 8624858 Review.
Cited by
-
Bullous pemphigoid following COVID-19 vaccine: An autoimmune disorder.Ann Med Surg (Lond). 2022 Aug;80:104266. doi: 10.1016/j.amsu.2022.104266. Epub 2022 Jul 31. Ann Med Surg (Lond). 2022. PMID: 35936564 Free PMC article. No abstract available.
-
Autoreactive Peripheral Blood T Helper Cell Responses in Bullous Pemphigoid and Elderly Patients With Pruritic Disorders.Front Immunol. 2021 Mar 25;12:569287. doi: 10.3389/fimmu.2021.569287. eCollection 2021. Front Immunol. 2021. PMID: 33841390 Free PMC article.
-
The pathogenesis of bullous skin diseases.J Transl Autoimmun. 2019 Aug 26;2:100014. doi: 10.1016/j.jtauto.2019.100014. eCollection 2019 Dec. J Transl Autoimmun. 2019. PMID: 32743502 Free PMC article. Review.
-
Junctional epithelium and hemidesmosomes: Tape and rivets for solving the "percutaneous device dilemma" in dental and other permanent implants.Bioact Mater. 2022 Mar 19;18:178-198. doi: 10.1016/j.bioactmat.2022.03.019. eCollection 2022 Dec. Bioact Mater. 2022. PMID: 35387164 Free PMC article. Review.
-
Anti-BP180 Autoantibodies Are Present in Stroke and Recognize Human Cutaneous BP180 and BP180-NC16A.Front Immunol. 2019 Feb 26;10:236. doi: 10.3389/fimmu.2019.00236. eCollection 2019. Front Immunol. 2019. PMID: 30863396 Free PMC article.
References
-
- Caca-Biljanovska N, Arsovska-Bezhoska I, V’Lckova-Laskoska M. PUVA-induced bullous pemphigoid in psoriasis. Acta Dermatovenerologica Croatica (2016) 24:214–7. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials