

High-resolution global peptide-protein docking using fragments-based PIPER-FlexPepDock
- PMID: 29281622
- PMCID: PMC5760072
- DOI: 10.1371/journal.pcbi.1005905
High-resolution global peptide-protein docking using fragments-based PIPER-FlexPepDock
Abstract
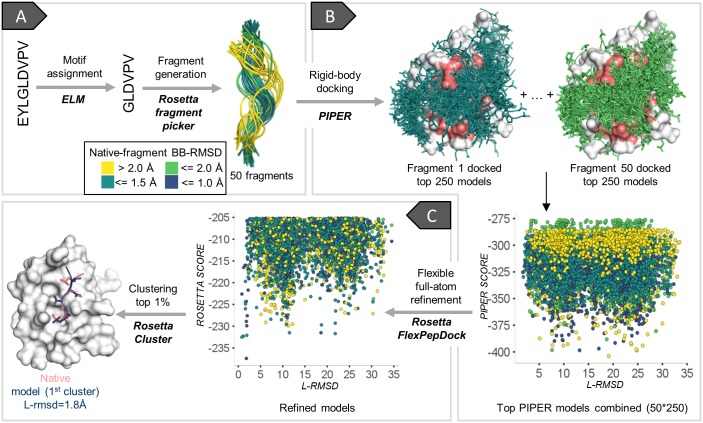
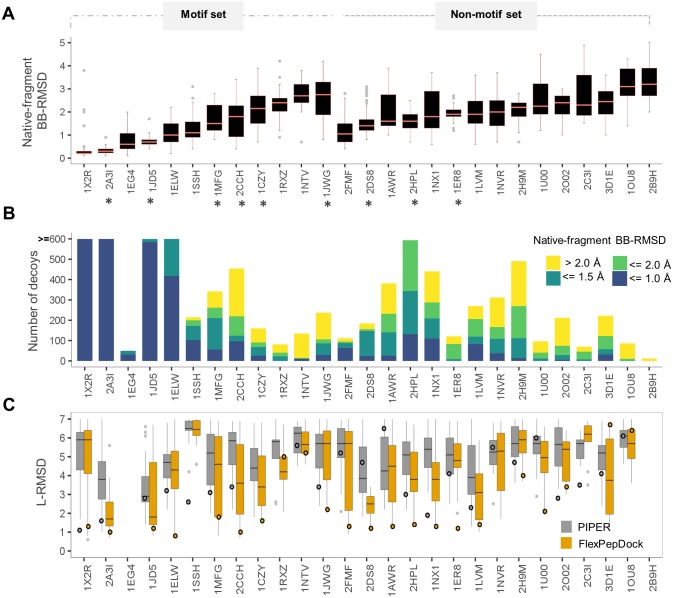
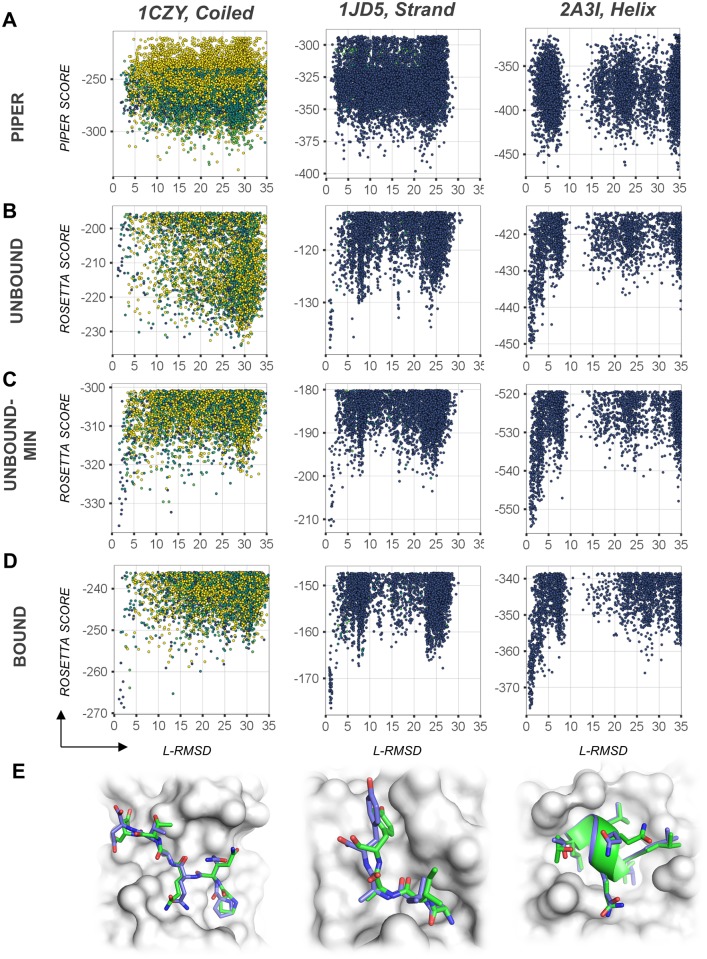
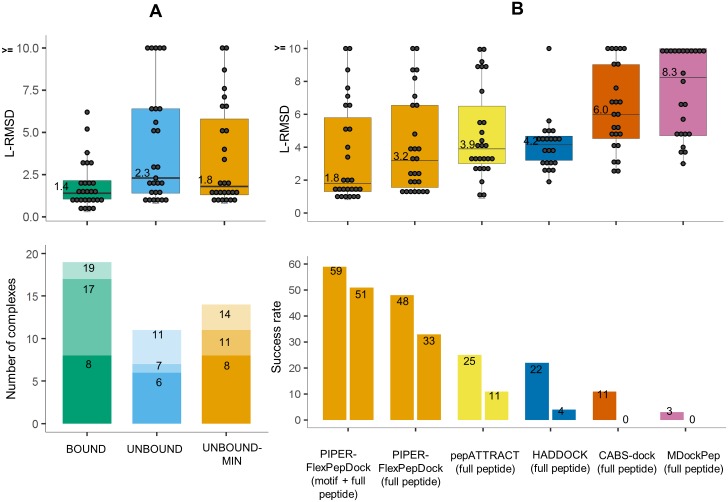
Peptide-protein interactions contribute a significant fraction of the protein-protein interactome. Accurate modeling of these interactions is challenging due to the vast conformational space associated with interactions of highly flexible peptides with large receptor surfaces. To address this challenge we developed a fragment based high-resolution peptide-protein docking protocol. By streamlining the Rosetta fragment picker for accurate peptide fragment ensemble generation, the PIPER docking algorithm for exhaustive fragment-receptor rigid-body docking and Rosetta FlexPepDock for flexible full-atom refinement of PIPER docked models, we successfully addressed the challenge of accurate and efficient global peptide-protein docking at high-resolution with remarkable accuracy, as validated on a small but representative set of peptide-protein complex structures well resolved by X-ray crystallography. Our approach opens up the way to high-resolution modeling of many more peptide-protein interactions and to the detailed study of peptide-protein association in general. PIPER-FlexPepDock is freely available to the academic community as a server at http://piperfpd.furmanlab.cs.huji.ac.il.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Pawson T, Nash P. Assembly of cell regulatory systems through protein interaction domains. Science. 2003;300(5618):445–52. doi: 10.1126/science.1083653 - DOI - PubMed
-
- Petsalaki E, Russell RB. Peptide-mediated interactions in biological systems: new discoveries and applications. Curr Opin Biotechnol. 2008;19(4):344–50. doi: 10.1016/j.copbio.2008.06.004 - DOI - PubMed
-
- Neduva V, Linding R, Su-Angrand I, Stark A, de Masi F, Gibson TJ, et al. Systematic discovery of new recognition peptides mediating protein interaction networks. PLoS Biol. 2005;3(12):e405 doi: 10.1371/journal.pbio.0030405 - DOI - PMC - PubMed
-
- Vacic V, Oldfield CJ, Mohan A, Radivojac P, Cortese MS, Uversky VN, et al. Characterization of molecular recognition features, MoRFs, and their binding partners. J Proteome Res. 2007;6(6):2351–66. doi: 10.1021/pr0701411 - DOI - PMC - PubMed
-
- Gamble TR, Vajdos FF, Yoo S, Worthylake DK, Houseweart M, Sundquist WI, et al. Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell. 1996;87(7):1285–94. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
