

Development of a Multilocus Sequence Typing (MLST) scheme for Treponema pallidum subsp. pertenue: Application to yaws in Lihir Island, Papua New Guinea
- PMID: 29281641
- PMCID: PMC5760108
- DOI: 10.1371/journal.pntd.0006113
Development of a Multilocus Sequence Typing (MLST) scheme for Treponema pallidum subsp. pertenue: Application to yaws in Lihir Island, Papua New Guinea
Abstract
Background: Yaws is a neglected tropical disease, caused by Treponema pallidum subsp. pertenue. The disease causes chronic lesions, primarily in young children living in remote villages in tropical climates. As part of a global yaws eradication campaign initiated by the World Health Organization, we sought to develop and evaluate a molecular typing method to distinguish different strains of T. pallidum subsp. pertenue for disease control and epidemiological purposes.
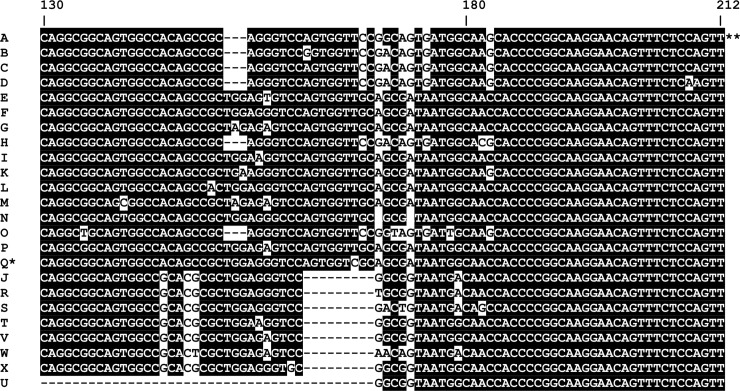
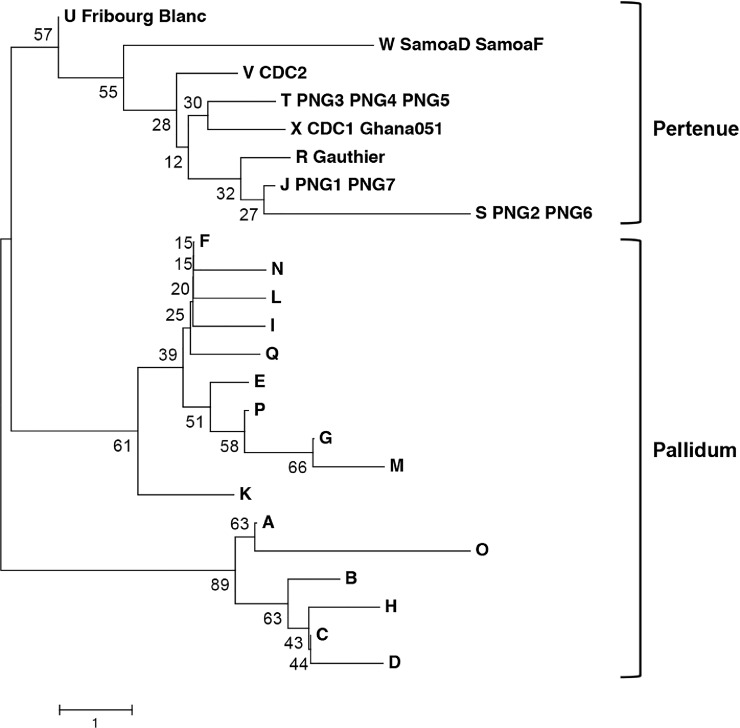
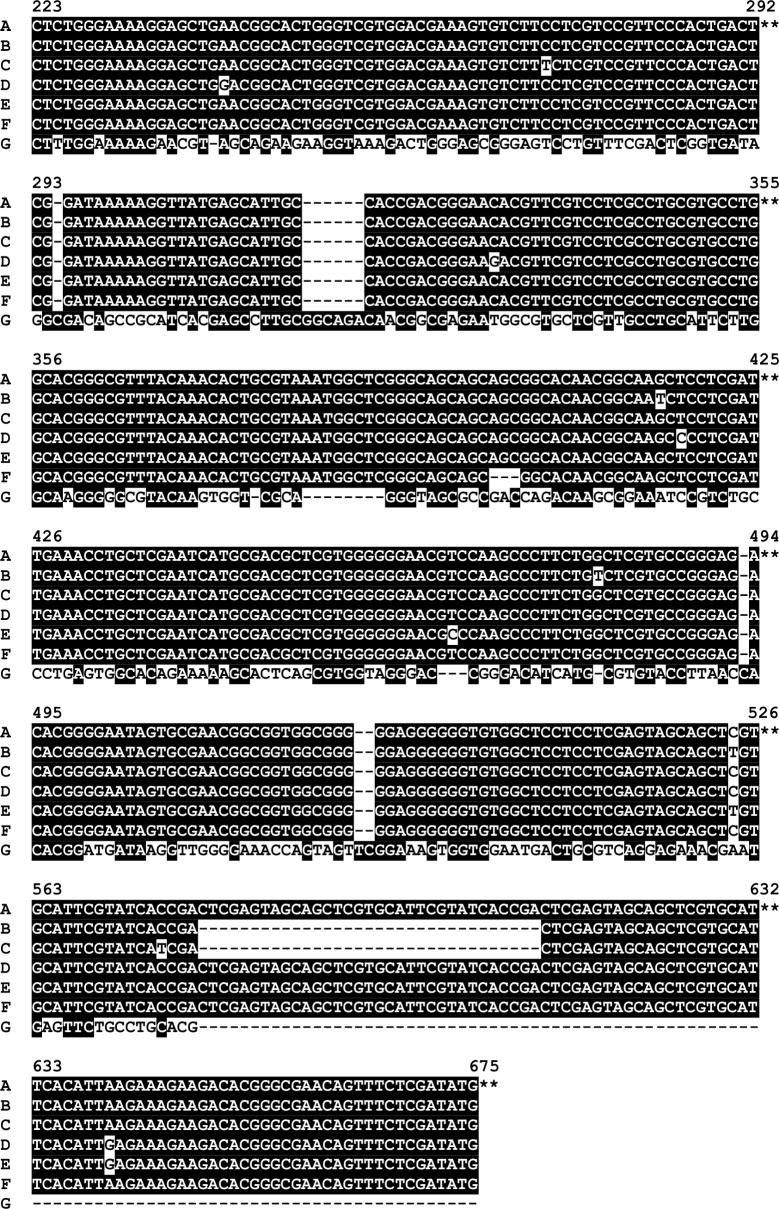
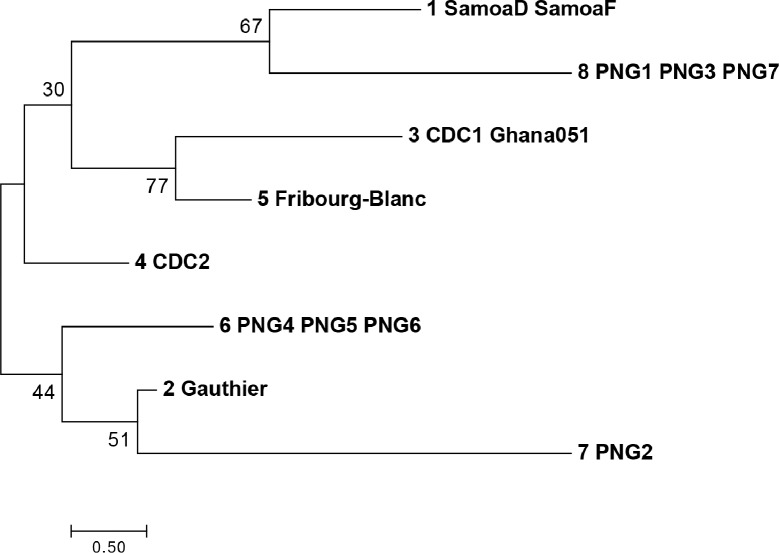
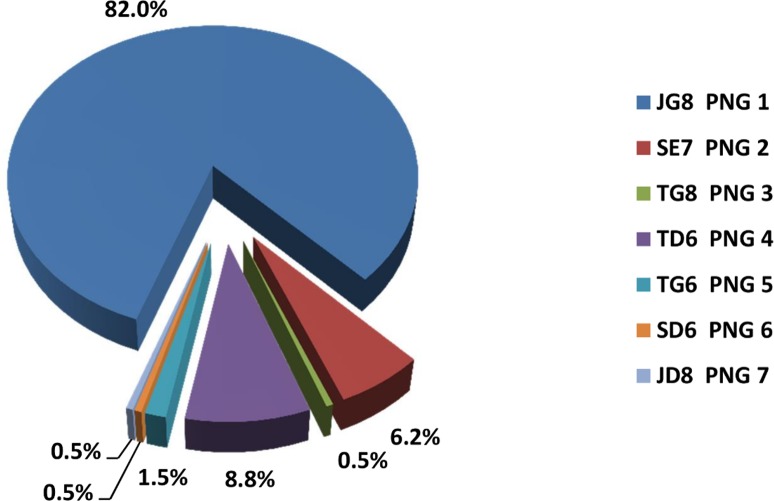
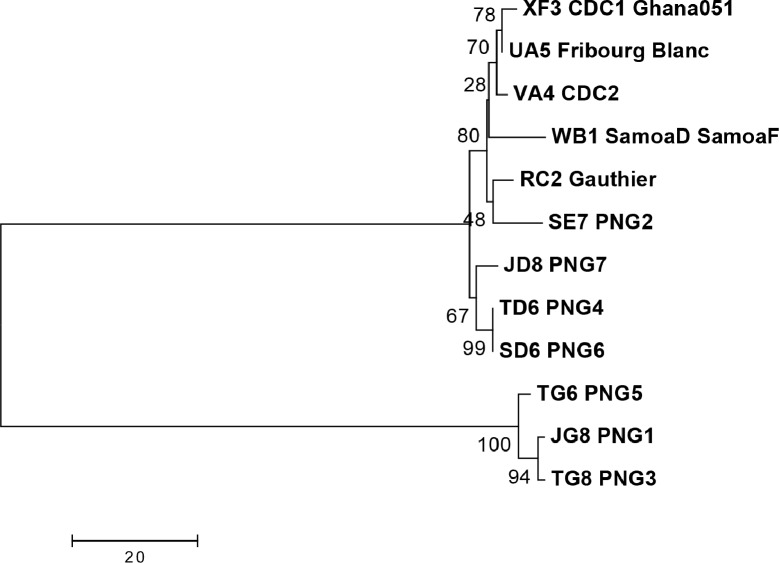
Methods and principal findings: Published genome sequences of strains of T. pallidum subsp. pertenue and pallidum were compared to identify polymorphic genetic loci among the strains. DNA from a number of existing historical Treponema isolates, as well as a subset of samples from yaws patients collected in Lihir Island, Papua New Guinea, were analyzed using these targets. From these data, three genes (tp0548, tp0136 and tp0326) were ultimately selected to give a high discriminating capability among the T. pallidum subsp. pertenue samples tested. Intragenic regions of these three target genes were then selected to enhance the discriminating capability of the typing scheme using short readily amplifiable loci. This 3-gene multilocus sequence typing (MLST) method was applied to existing historical human yaws strains, the Fribourg-Blanc simian isolate, and DNA from 194 lesion swabs from yaws patients on Lihir Island, Papua New Guinea. Among all samples tested, fourteen molecular types were identified, seven of which were found in patient samples and seven among historical isolates or DNA. Three types (JG8, TD6, and SE7) were predominant on Lihir Island.
Conclusions: This MLST approach allows molecular typing and differentiation of yaws strains. This method could be a useful tool to complement epidemiological studies in regions where T. pallidum subsp. pertenue is prevalent with the overall goals of improving our understanding of yaws transmission dynamics and helping the yaws eradication campaign to succeed.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures



References
-
- Antal GM, Lukehart SA, Meheus AZ. The Endemic Treponematoses. Microbes Infect. 2002;4: 83–94. - PubMed
-
- Giacani L, Lukehart SA. The Endemic Treponematoses. Clin Microbiol Rev. 2014;27: 89–115. doi: 10.1128/CMR.00070-13 - DOI - PMC - PubMed
-
- Asiedu K, Fitzpatrick C, Jannin J. Eradication of Yaws: Historical Efforts and Achieving WHO’s 2020 Target. PLoS Negl Trop Dis. 2014;8: e3016 doi: 10.1371/journal.pntd.0003016 - DOI - PMC - PubMed
-
- Čejková D, Zobaníková M, Chen L, Pospíšilová P, Strouhal M, Qin X, et al. Whole Genome Sequences of Three Treponema pallidum ssp. pertenue Strains: Yaws and Syphilis Treponemes Differ in Less than 0.2% of the Genome Sequence. PLoS Negl Trop Dis. 2012;6: e1471 doi: 10.1371/journal.pntd.0001471 - DOI - PMC - PubMed
-
- Mikalová L, Strouhal M, Čejková D, Zobaníková M, Pospíšilová P, Norris SJ, et al. Genome Analysis of Treponema pallidum subsp. pallidum and subsp. pertenue Strains: Most of the Genetic Differences Are Localized in Six Regions. PLoS ONE. 2010;5: e15713 doi: 10.1371/journal.pone.0015713 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
