

Somatic Mutations Activating the mTOR Pathway in Dorsal Telencephalic Progenitors Cause a Continuum of Cortical Dysplasias
- PMID: 29281825
- PMCID: PMC5752134
- DOI: 10.1016/j.celrep.2017.11.106
Somatic Mutations Activating the mTOR Pathway in Dorsal Telencephalic Progenitors Cause a Continuum of Cortical Dysplasias
Abstract
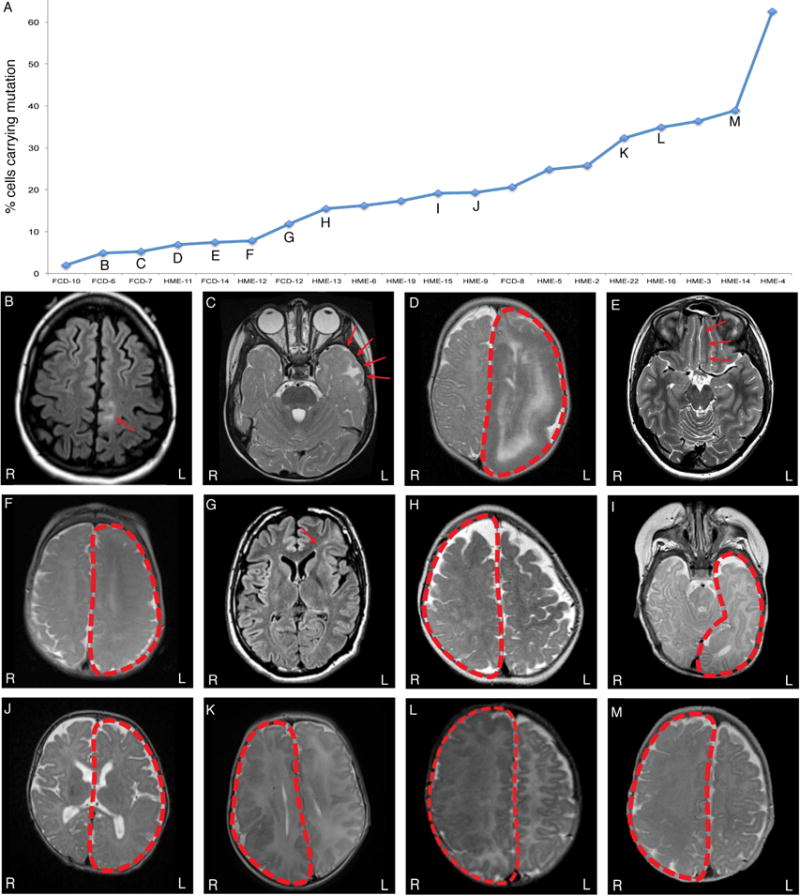
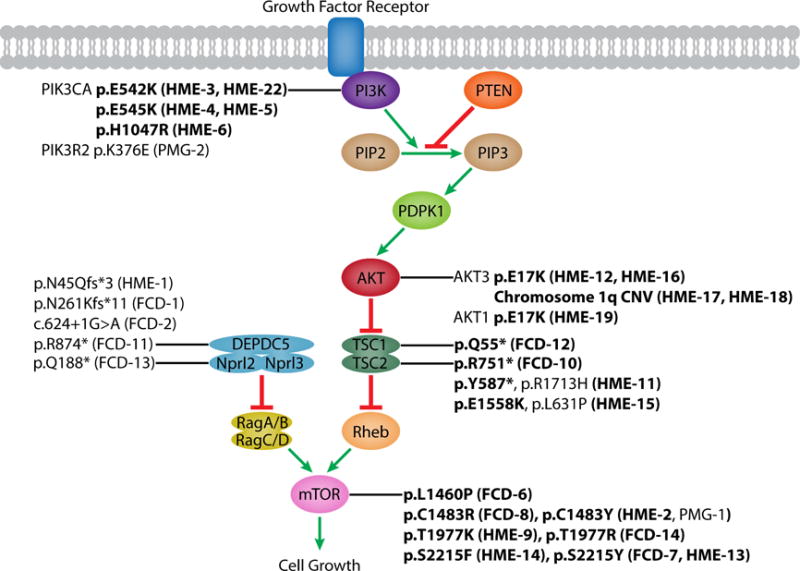
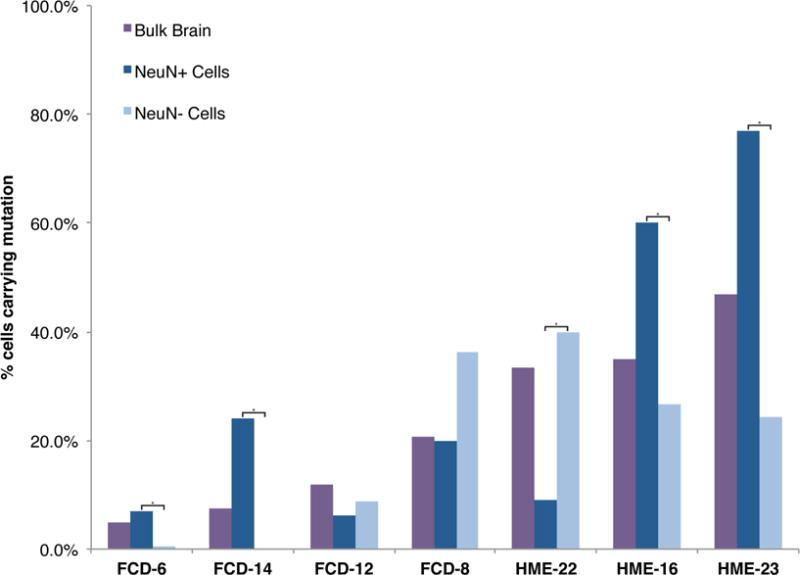
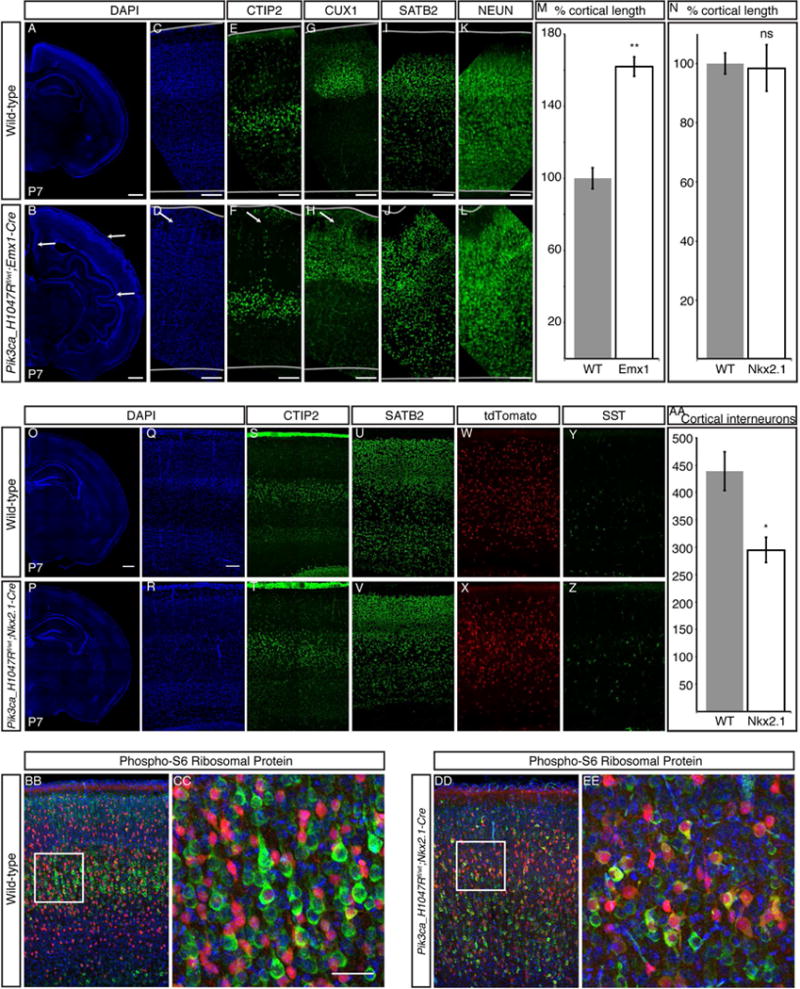
Focal cortical dysplasia (FCD) and hemimegalencephaly (HME) are epileptogenic neurodevelopmental malformations caused by mutations in mTOR pathway genes. Deep sequencing of these genes in FCD/HME brain tissue identified an etiology in 27 of 66 cases (41%). Radiographically indistinguishable lesions are caused by somatic activating mutations in AKT3, MTOR, and PIK3CA and germline loss-of-function mutations in DEPDC5, NPRL2, and TSC1/2, including TSC2 mutations in isolated HME demonstrating a "two-hit" model. Mutations in the same gene cause a disease continuum from FCD to HME to bilateral brain overgrowth, reflecting the progenitor cell and developmental time when the mutation occurred. Single-cell sequencing demonstrated mTOR activation in neurons in all lesions. Conditional Pik3ca activation in the mouse cortex showed that mTOR activation in excitatory neurons and glia, but not interneurons, is sufficient for abnormal cortical overgrowth. These data suggest that mTOR activation in dorsal telencephalic progenitors, in some cases specifically the excitatory neuron lineage, causes cortical dysplasia.
Keywords: brain malformations; cortical development; epilepsy; excitatory neurons; focal cortical dysplasia; hemimegalancephaly; mTOR pathway; next-generation sequencing; single-cell sequencing; somatic mutations.
Copyright © 2017 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
H.V.V. owns stock in several pharmaceutical companies and has received honoraria for lectures (no direct conflict with this paper). G.W.M. serves on the Data Management Committee for Neuropace, Inc. I.B. has received honoraria for lectures (no direct conflict with this paper).
Figures
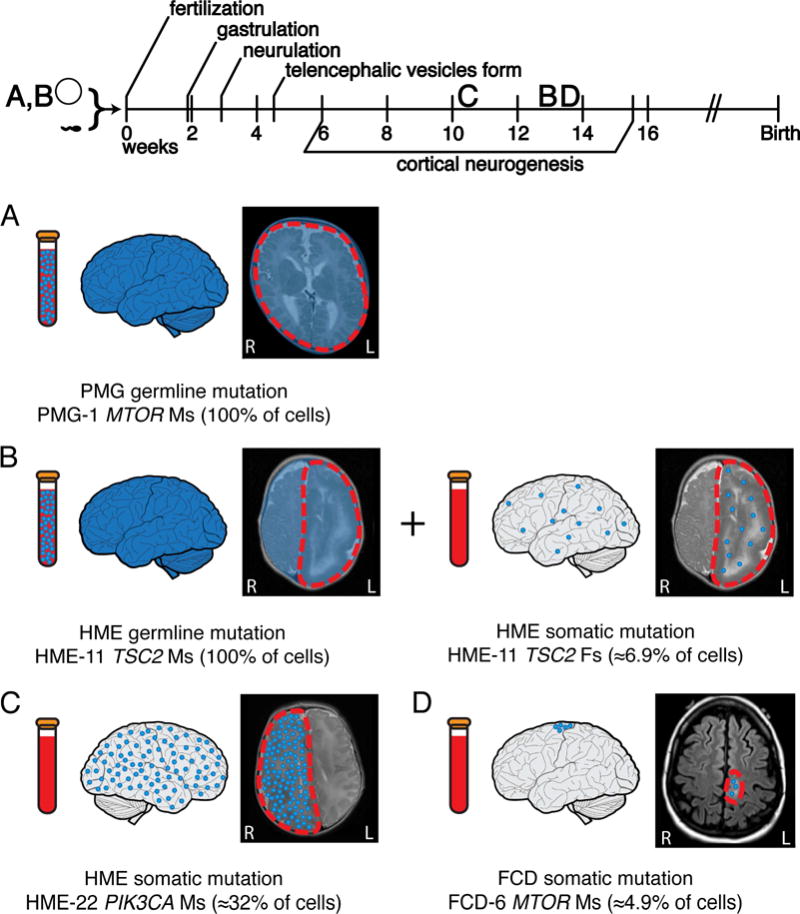
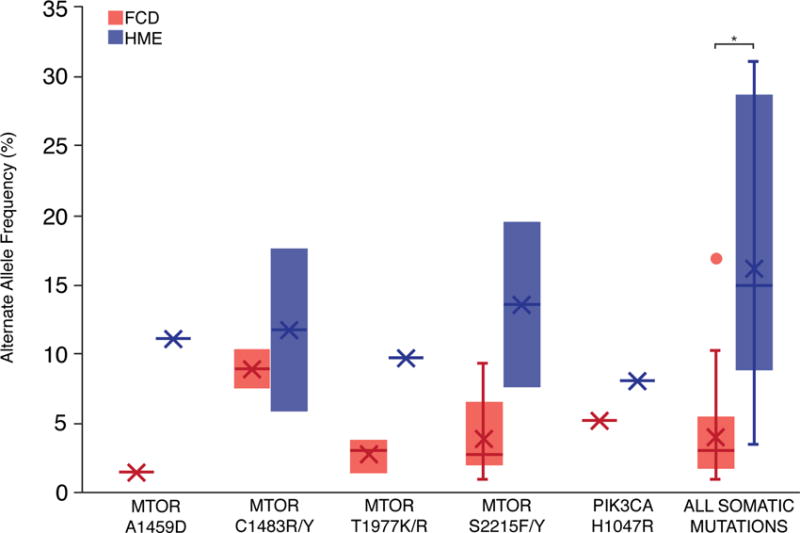
Comment in
-
The Space-Time Continuum of Cortical Dysplasia.Epilepsy Curr. 2019 Jan;19(1):54-56. doi: 10.1177/1535759718822039. Epub 2019 Jan 30. Epilepsy Curr. 2019. PMID: 30838928 Free PMC article.
References
-
- Baulac S, Ishida S, Marsan E, Miquel C, Biraben A, Nguyen DK, Nordli D, Cossette P, Nguyen S, Lambrecq V, et al. Familial focal epilepsy with focal cortical dysplasia due to DEPDC5 mutations. Annals of neurology. 2015;77:675–683. - PubMed
-
- Blumcke I, Sarnat HB. Somatic mutations rather than viral infection classify focal cortical dysplasia type II as mTORopathy. Curr Opin Neurol. 2016;29:388–395. - PubMed
-
- Blumcke I, Spreafico R, Haaker G, Coras R, Kobow K, Bien CG, Pfafflin M, Elger C, Widman G, Schramm J, et al. Histopathological Findings in Brain Tissue Obtained during Epilepsy Surgery. N Engl J Med. 2017;377:1648–1656. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 NS032457/NS/NINDS NIH HHS/United States
- U01 MH106883/MH/NIMH NIH HHS/United States
- T32 GM007226/GM/NIGMS NIH HHS/United States
- T32 GM007753/GM/NIGMS NIH HHS/United States
- R01 NS079277/NS/NINDS NIH HHS/United States
- P30 HD018655/HD/NICHD NIH HHS/United States
- R01 NS038992/NS/NINDS NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- U54 HD090255/HD/NICHD NIH HHS/United States
- R01 NS083823/NS/NINDS NIH HHS/United States
- K23 NS069784/NS/NINDS NIH HHS/United States
- R01 NS035129/NS/NINDS NIH HHS/United States
- P50 CA211015/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
