

Within-host evolution of Enterococcus faecium during longitudinal carriage and transition to bloodstream infection in immunocompromised patients
- PMID: 29282103
- PMCID: PMC5744393
- DOI: 10.1186/s13073-017-0507-0
Within-host evolution of Enterococcus faecium during longitudinal carriage and transition to bloodstream infection in immunocompromised patients
Abstract
Background: Enterococcus faecium is a leading cause of hospital-acquired infection, particularly in the immunocompromised. Here, we use whole genome sequencing of E. faecium to study within-host evolution and the transition from gut carriage to invasive disease.
Methods: We isolated and sequenced 180 E. faecium from four immunocompromised patients who developed bloodstream infection during longitudinal surveillance of E. faecium in stool and their immediate environment.
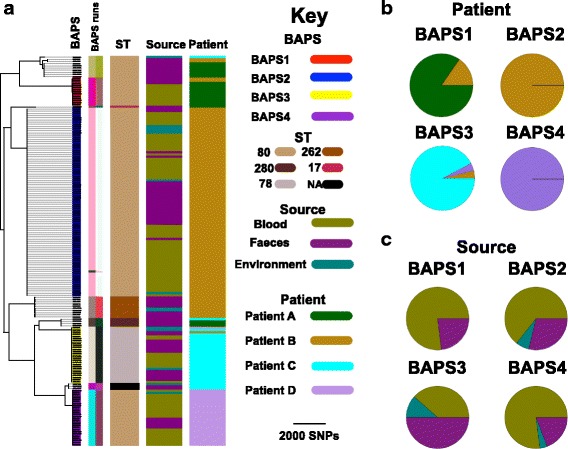
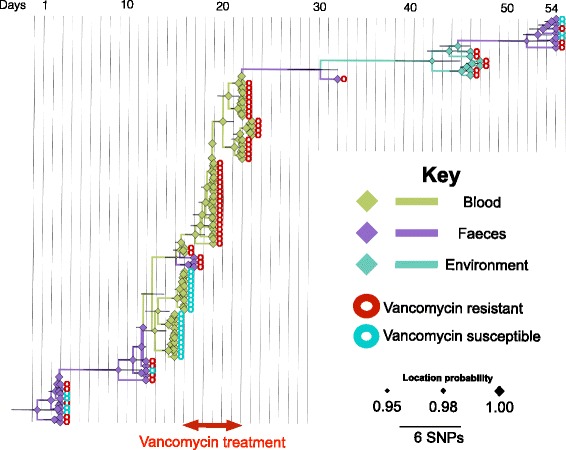
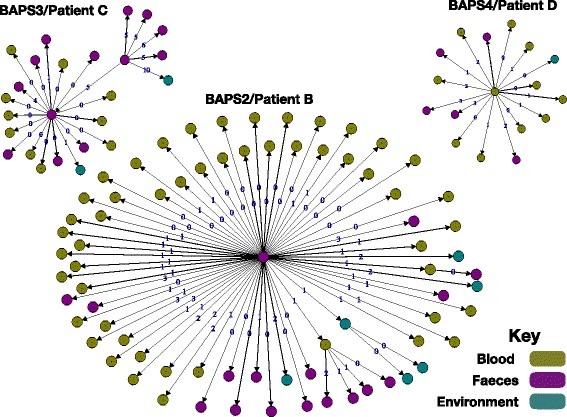
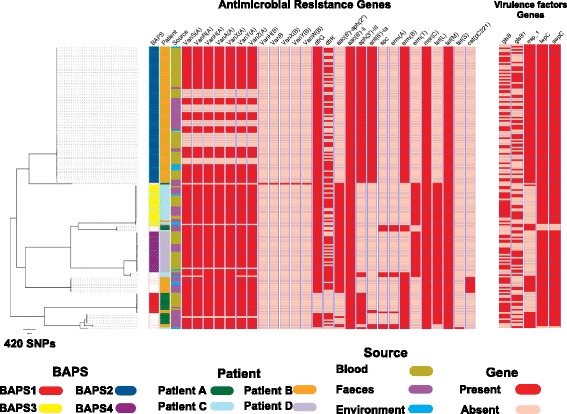
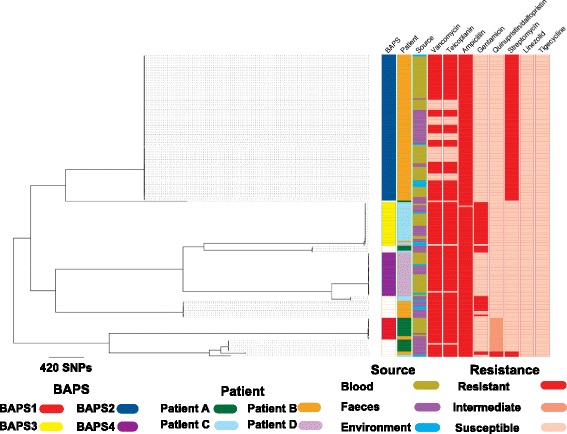
Results: A phylogenetic tree based on single nucleotide polymorphisms (SNPs) in the core genome of the 180 isolates demonstrated several distinct clones. This was highly concordant with the population structure inferred by Bayesian methods, which contained four main BAPS (Bayesian Analysis of Population Structure) groups. The majority of isolates from each patient resided in a single group, but all four patients also carried minority populations in stool from multiple phylogenetic groups. Bloodstream isolates from each case belonged to a single BAPS group, which differed in all four patients. Analysis of 87 isolates (56 from blood) belonging to a single BAPS group that were cultured from the same patient over 54 days identified 30 SNPs in the core genome (nine intergenic, 13 non-synonymous, eight synonymous), and 250 accessory genes that were variably present. Comparison of these genetic variants in blood isolates versus those from stool or environment did not identify any variants associated with bloodstream infection. The substitution rate for these isolates was estimated to be 128 (95% confidence interval 79.82 181.77) mutations per genome per year, more than ten times higher than previous estimates for E. faecium. Within-patient variation in vancomycin resistance associated with vanA was common and could be explained by plasmid loss, or less often by transposon loss.
Conclusions: These findings demonstrate the diversity of E. faecium carriage by individual patients and significant within-host diversity of E. faecium, but do not provide evidence for adaptive genetic variation associated with invasion.
Keywords: Enterococcus faecium; Genome sequencing; Within-host evolution.
Conflict of interest statement
Ethics approval and consent to participate
The study was approved by the CUH Research and Development Department (reference A093285) and the National Research Ethics Service East of England Ethics Committee (reference 14/EE/1123). Written informed consent was obtained from all participants prior to enrolment to the study. All human subjects were adult. We declare that the study conformed to the Declaration of Helsinki.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
Similar articles
-
Genetic relatedness and risk factor analysis of ampicillin-resistant and high-level gentamicin-resistant enterococci causing bloodstream infections in Tanzanian children.BMC Infect Dis. 2015 Feb 28;15:107. doi: 10.1186/s12879-015-0845-8. BMC Infect Dis. 2015. PMID: 25884316 Free PMC article.
-
Complex Routes of Nosocomial Vancomycin-Resistant Enterococcus faecium Transmission Revealed by Genome Sequencing.Clin Infect Dis. 2017 Apr 1;64(7):886-893. doi: 10.1093/cid/ciw872. Clin Infect Dis. 2017. PMID: 28362945 Free PMC article.
-
Genomic Surveillance of Enterococcus faecium Reveals Limited Sharing of Strains and Resistance Genes between Livestock and Humans in the United Kingdom.mBio. 2018 Nov 6;9(6):e01780-18. doi: 10.1128/mBio.01780-18. mBio. 2018. PMID: 30401778 Free PMC article.
-
A decade of genomic history for healthcare-associated Enterococcus faecium in the United Kingdom and Ireland.Genome Res. 2016 Oct;26(10):1388-1396. doi: 10.1101/gr.204024.116. Epub 2016 Aug 15. Genome Res. 2016. PMID: 27527616 Free PMC article.
-
Restricted gene flow among hospital subpopulations of Enterococcus faecium.mBio. 2012 Jul 17;3(4):e00151-12. doi: 10.1128/mBio.00151-12. Print 2012. mBio. 2012. PMID: 22807567 Free PMC article.
Cited by
-
Introduction and spread of vancomycin-resistant Enterococcus faecium (VREfm) at a German tertiary care medical center from 2004 until 2010: a retrospective whole-genome sequencing (WGS) study of the molecular epidemiology of VREfm.Antimicrob Resist Infect Control. 2024 Feb 14;13(1):20. doi: 10.1186/s13756-024-01379-4. Antimicrob Resist Infect Control. 2024. PMID: 38355509 Free PMC article.
-
Enterococcus faecium genome dynamics during long-term asymptomatic patient gut colonization.Microb Genom. 2019 Jul;5(7):e000277. doi: 10.1099/mgen.0.000277. Epub 2019 Jun 5. Microb Genom. 2019. PMID: 31166888 Free PMC article.
-
A core genome approach that enables prospective and dynamic monitoring of infectious outbreaks.Sci Rep. 2019 May 24;9(1):7808. doi: 10.1038/s41598-019-44189-0. Sci Rep. 2019. PMID: 31127153 Free PMC article.
-
Genome and transcriptome analysis of Enterococcus faecium from intestinal colonization and Enterococcus faecium from urinary tract infection.Front Microbiol. 2023 Oct 31;14:1273949. doi: 10.3389/fmicb.2023.1273949. eCollection 2023. Front Microbiol. 2023. PMID: 38029192 Free PMC article.
-
Replicative selfish genetic elements are driving rapid pathogenic adaptation of Enterococcus faecium.bioRxiv [Preprint]. 2025 Mar 16:2025.03.16.643550. doi: 10.1101/2025.03.16.643550. bioRxiv. 2025. PMID: 40161577 Free PMC article. Preprint.
References
-
- McCracken M, Wong A, Mitchell R, Gravel D, Conly J, Embil J, et al. Molecular epidemiology of vancomycin-resistant enterococcal bacteraemia: results from the Canadian Nosocomial Infection Surveillance Program, 1999-2009. J Antimicrob Chemother. 2013;68(7):1505–9. doi: 10.1093/jac/dkt054. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
