

Regulation of redox balance in cancer and T cells
- PMID: 29282291
- PMCID: PMC5961053
- DOI: 10.1074/jbc.TM117.000257
Regulation of redox balance in cancer and T cells
Abstract
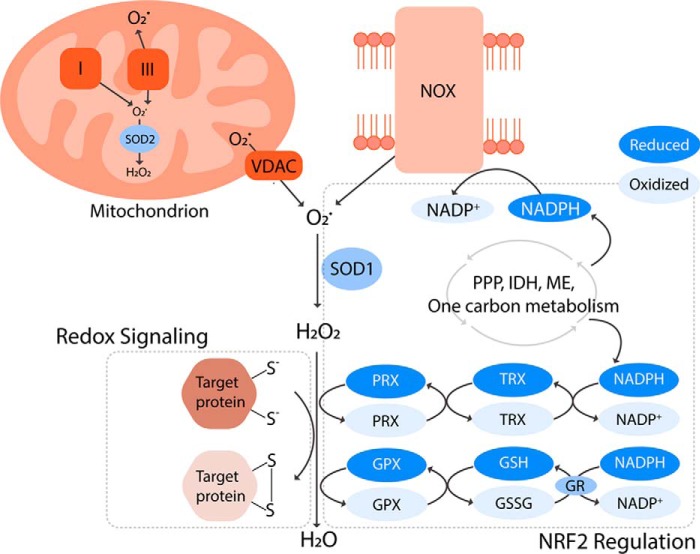
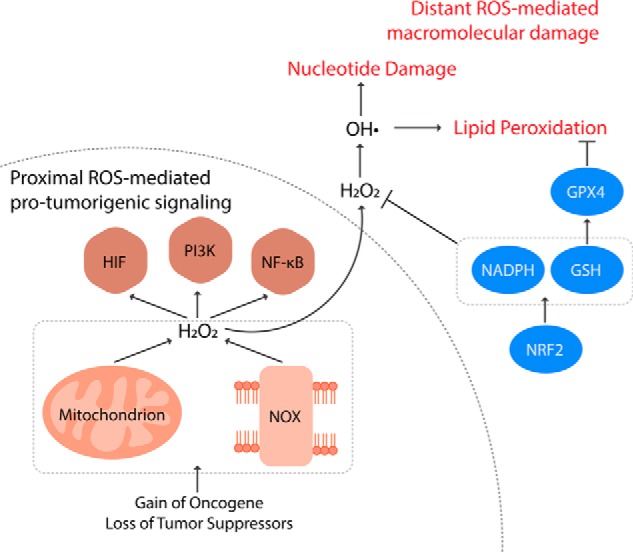
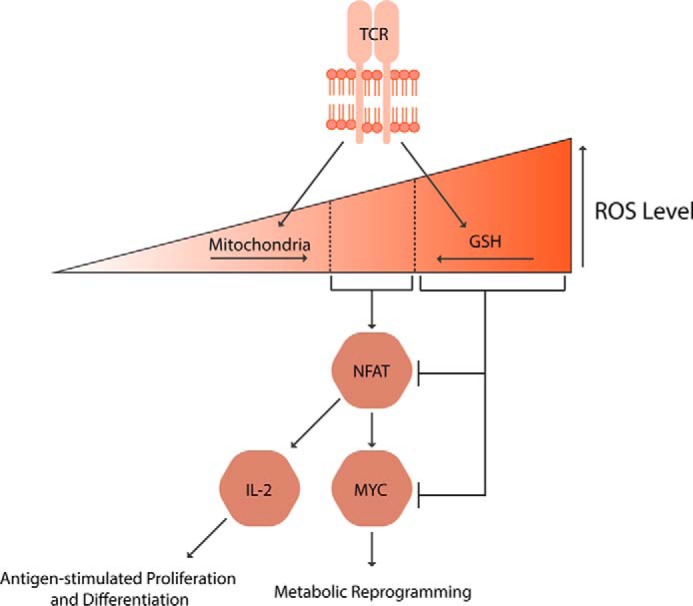
Reactive oxygen species (ROS) mediate redox signaling necessary for numerous cellular functions. Yet, high levels of ROS in cells and tissues can cause damage and cell death. Therefore, regulation of redox homeostasis is essential for ROS-dependent signaling that does not incur cellular damage. Cells achieve this optimal balance by coordinating ROS production and elimination. In this Minireview, we discuss the mechanisms by which proliferating cancer and T cells maintain a carefully controlled redox balance. Greater insight into such redox biology may enable precisely targeted manipulation of ROS for effective medical therapies against cancer or immunological disorders.
Keywords: T-cell; cancer; reactive oxygen species (ROS); redox regulation; redox signaling.
© 2018 by The American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
References
-
- Chouchani E. T., Pell V. R., Gaude E., Aksentijević D., Sundier S. Y., Robb E. L., Logan A., Nadtochiy S. M., Ord E. N. J., Smith A. C., Eyassu F., Shirley R., Hu C. H., Dare A. J., James A. M., et al. (2014) Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435 10.1038/nature13909 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
