

Acid sphingomyelinase promotes mitochondrial dysfunction due to glutamate-induced regulated necrosis
- PMID: 29282302
- PMCID: PMC5794425
- DOI: 10.1194/jlr.M080374
Acid sphingomyelinase promotes mitochondrial dysfunction due to glutamate-induced regulated necrosis
Abstract
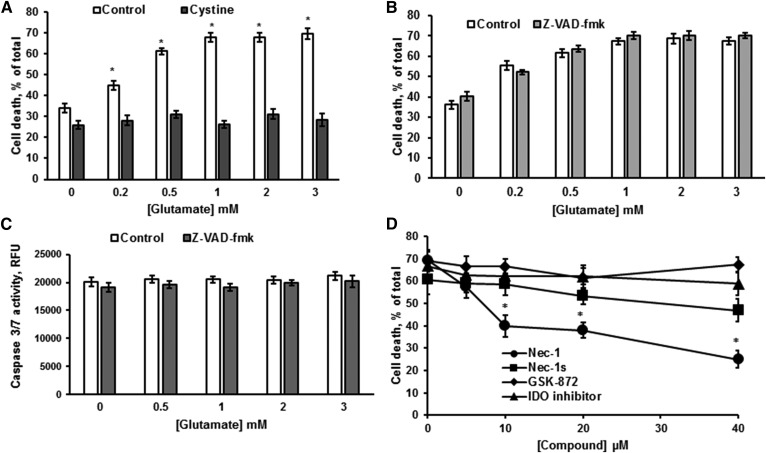
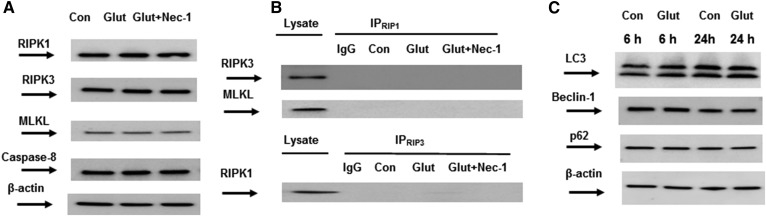
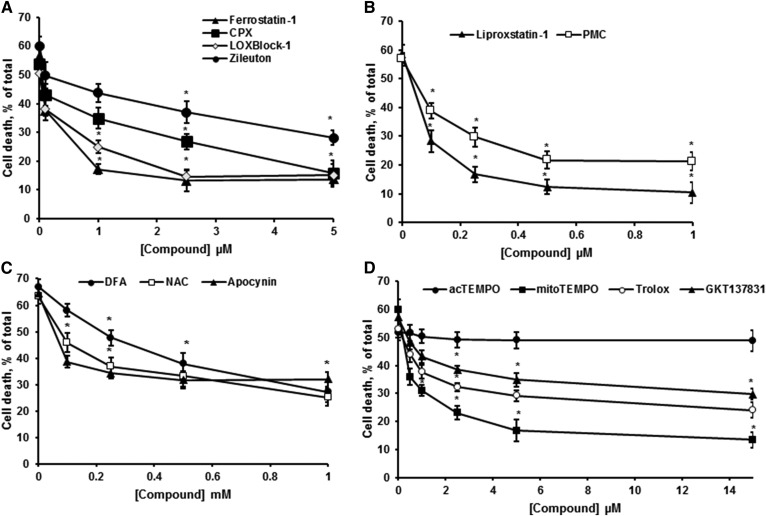
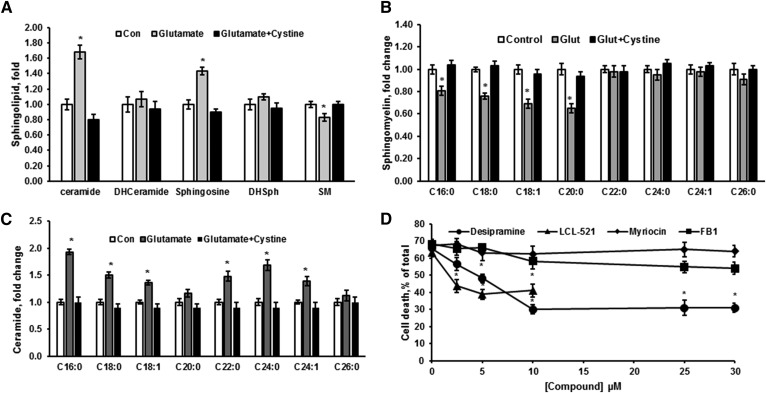
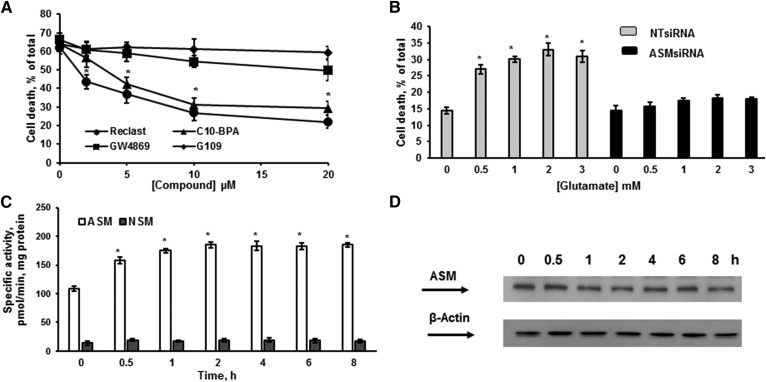
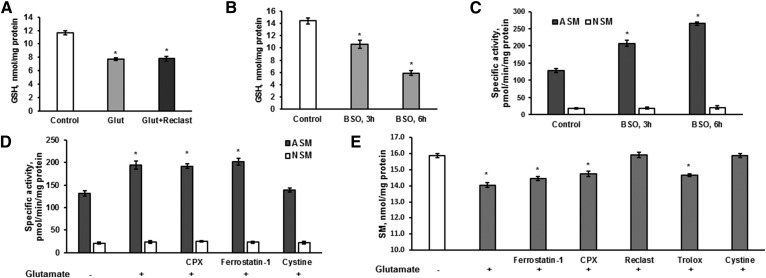
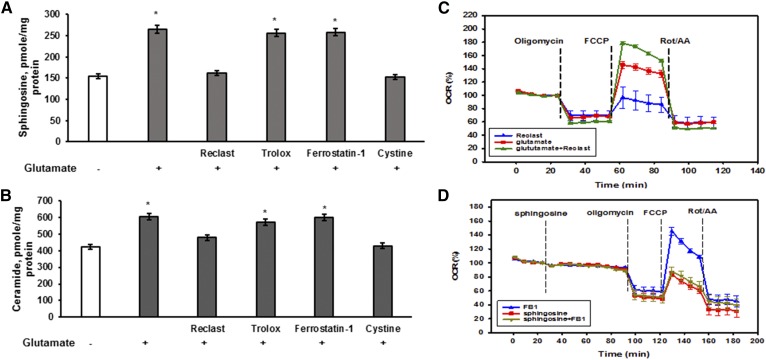
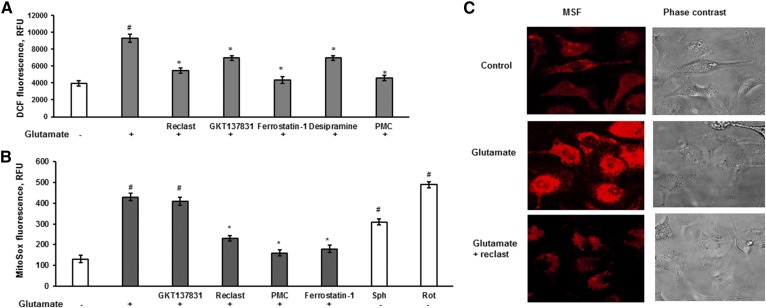
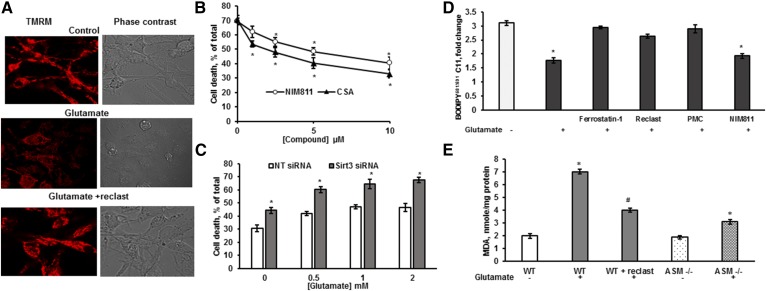
Inhibiting the glutamate/cystine antiporter system xc-, a key antioxidant defense machinery in the CNS, could trigger a novel form of regulated necrotic cell death, ferroptosis. The underlying mechanisms of system xc--dependent cell demise were elucidated using primary oligodendrocytes (OLs) treated with glutamate to block system xc- function. Pharmacological analysis revealed ferroptosis as a major contributing factor to glutamate-initiated OL death. A sphingolipid profile showed elevations of ceramide species and sphingosine that were preventable by inhibiting of an acid sphingomyelinase (ASM) activity. OL survival was enhanced by both downregulating ASM expression and blocking ASM activity. Glutamate-induced ASM activation seems to involve posttranscriptional mechanisms and was associated with a decreased GSH level. Further investigation of the mechanisms of OL response to glutamate revealed enhanced reactive oxygen species production, augmented lipid peroxidation, and opening of the mitochondrial permeability transition pore that were attenuated by hindering ASM. Of note, knocking down sirtuin 3, a deacetylase governing the mitochondrial antioxidant system, reduced OL survival. The data highlight the importance of the mitochondrial compartment in regulated necrotic cell death and accentuate the novel role of ASM in disturbing mitochondrial functions during OL response to glutamate toxicity, which is essential for pathobiology in stroke and traumatic brain injury.
Keywords: brain lipids; ferroptosis; glutamate/cystine antiporter; lipids/peroxidation; mitochondria; oligodendrocytes; sirtuin; sphingolipids.
Copyright © 2018 by the American Society for Biochemistry and Molecular Biology, Inc.
Figures
Similar articles
-
Developmentally regulated ceramide synthase 6 increases mitochondrial Ca2+ loading capacity and promotes apoptosis.J Biol Chem. 2011 Feb 11;286(6):4644-58. doi: 10.1074/jbc.M110.164392. Epub 2010 Dec 10. J Biol Chem. 2011. PMID: 21148554 Free PMC article.
-
Oligodendrocyte excitotoxicity determined by local glutamate accumulation and mitochondrial function.J Neurochem. 2006 Jul;98(1):213-22. doi: 10.1111/j.1471-4159.2006.03861.x. Epub 2006 Apr 5. J Neurochem. 2006. PMID: 16606353
-
System xc- and glutamate transporter inhibition mediates microglial toxicity to oligodendrocytes.J Immunol. 2007 May 15;178(10):6549-56. doi: 10.4049/jimmunol.178.10.6549. J Immunol. 2007. PMID: 17475885
-
Molecular mechanisms and pathophysiology of necrotic cell death.Curr Mol Med. 2008 May;8(3):207-20. doi: 10.2174/156652408784221306. Curr Mol Med. 2008. PMID: 18473820 Review.
-
The role of acid sphingomyelinase and modulation of sphingolipid metabolism in bacterial infection.Biol Chem. 2018 Sep 25;399(10):1135-1146. doi: 10.1515/hsz-2018-0200. Biol Chem. 2018. PMID: 29924727 Review.
Cited by
-
Acid Sphingomyelinase, a Lysosomal and Secretory Phospholipase C, Is Key for Cellular Phospholipid Catabolism.Int J Mol Sci. 2021 Aug 20;22(16):9001. doi: 10.3390/ijms22169001. Int J Mol Sci. 2021. PMID: 34445706 Free PMC article. Review.
-
Novel Ferrocene Derivatives Induce Apoptosis through Mitochondria-Dependent and Cell Cycle Arrest via PI3K/Akt/mTOR Signaling Pathway in T Cell Acute Lymphoblastic Leukemia.Cancers (Basel). 2021 Sep 18;13(18):4677. doi: 10.3390/cancers13184677. Cancers (Basel). 2021. PMID: 34572904 Free PMC article.
-
Emerging Role of Ferroptosis in the Pathogenesis of Ischemic Stroke: A New Therapeutic Target?ASN Neuro. 2021 Jan-Dec;13:17590914211037505. doi: 10.1177/17590914211037505. ASN Neuro. 2021. PMID: 34463559 Free PMC article. Review.
-
Homozygous Smpd1 deficiency aggravates brain ischemia/ reperfusion injury by mechanisms involving polymorphonuclear neutrophils, whereas heterozygous Smpd1 deficiency protects against mild focal cerebral ischemia.Basic Res Cardiol. 2020 Oct 14;115(6):64. doi: 10.1007/s00395-020-00823-x. Basic Res Cardiol. 2020. PMID: 33057972 Free PMC article.
-
Pathogenesis of α-Synuclein in Parkinson's Disease: From a Neuron-Glia Crosstalk Perspective.Int J Mol Sci. 2022 Nov 25;23(23):14753. doi: 10.3390/ijms232314753. Int J Mol Sci. 2022. PMID: 36499080 Free PMC article. Review.
References
-
- Degterev A., Huang Z., Boyce M., Li Y., Jagtap P., Mizushima N., Cuny G. D., Mitchison T. J., Moskowitz M. A., and Yuan J.. 2005. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 1: 112–119. - PubMed
-
- Benveniste H., Drejer J., Schousboe A., and Diemer N.. 1984. Elevation of extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J. Neurochem. 43: 1369–1374. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
