

Enhanced anti-hepatocarcinoma efficacy by GLUT1 targeting and cellular microenvironment-responsive PAMAM-camptothecin conjugate
- PMID: 29282992
- PMCID: PMC6058575
- DOI: 10.1080/10717544.2017.1419511
Enhanced anti-hepatocarcinoma efficacy by GLUT1 targeting and cellular microenvironment-responsive PAMAM-camptothecin conjugate
Abstract
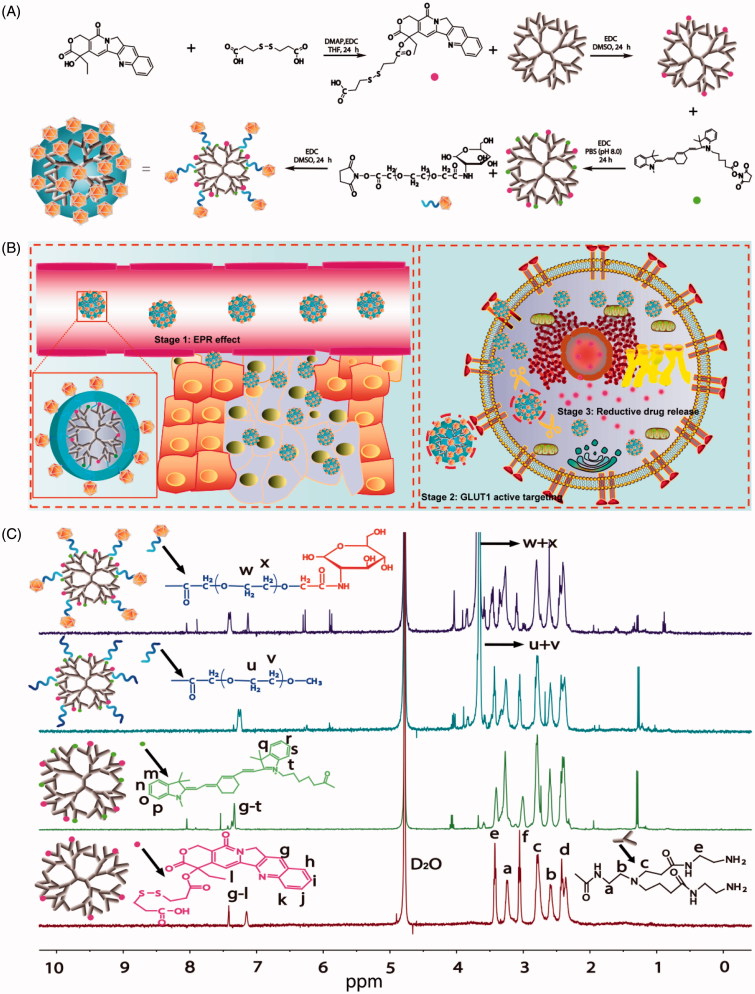
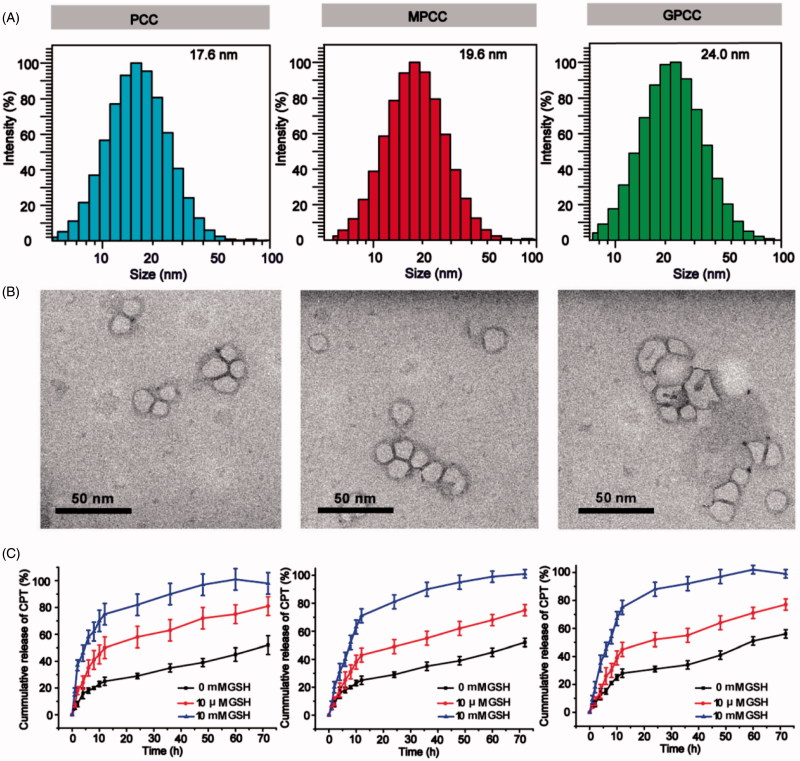
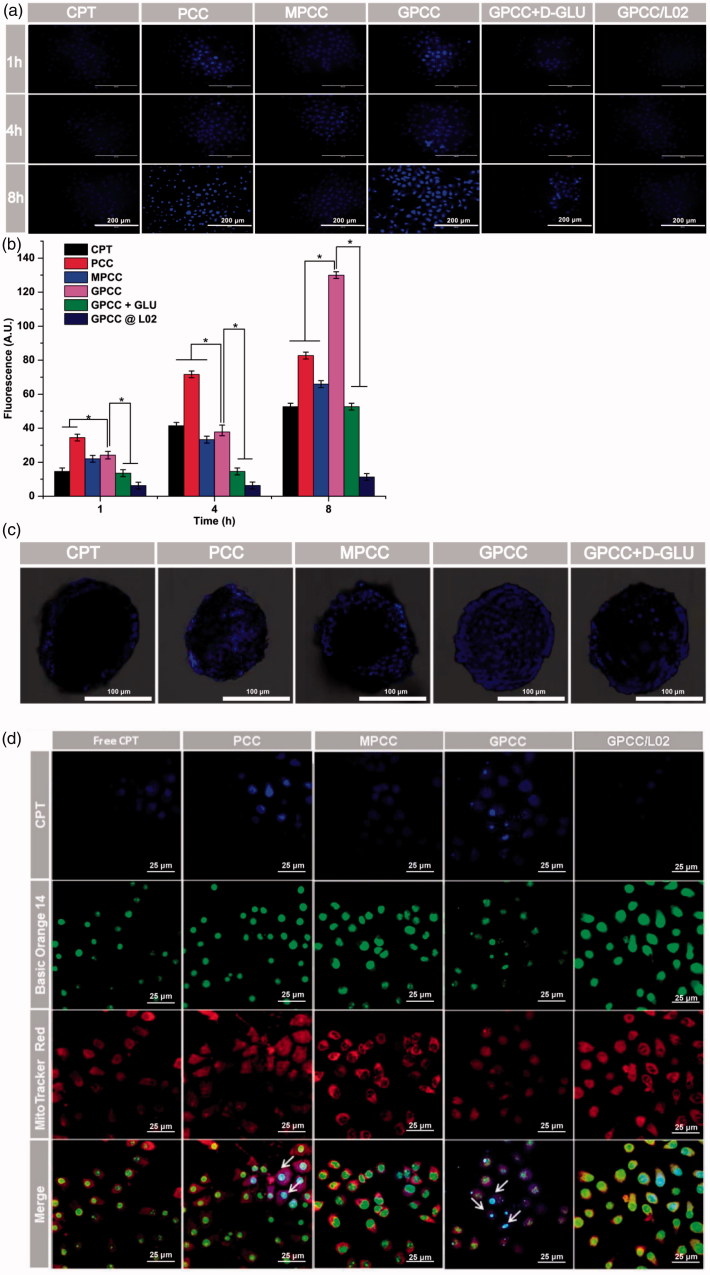
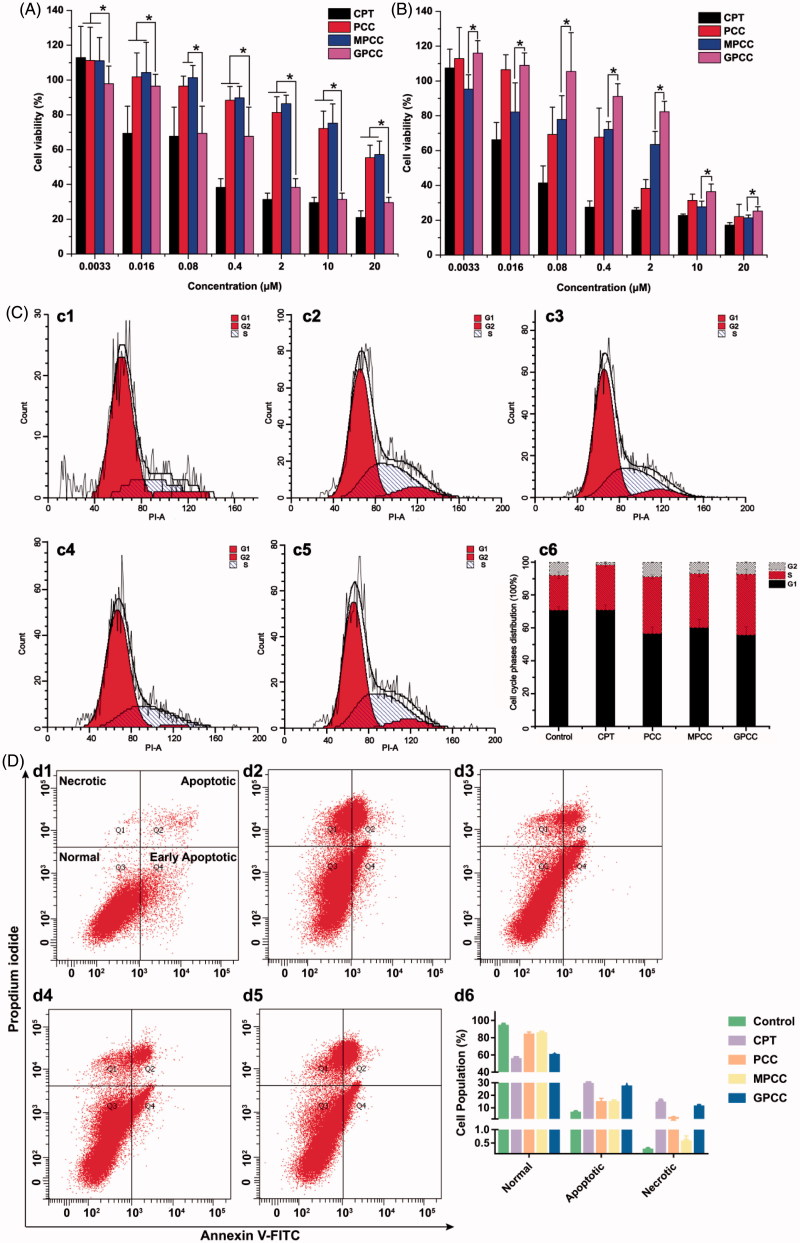
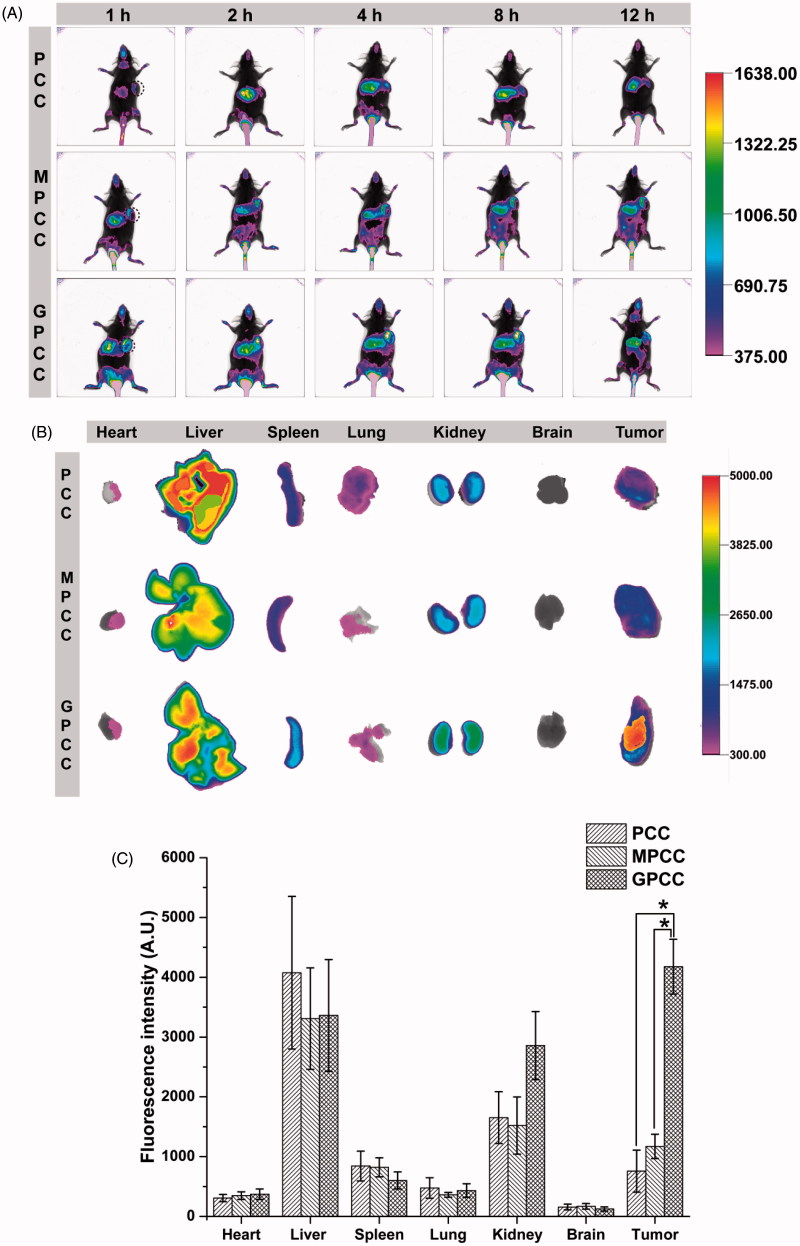
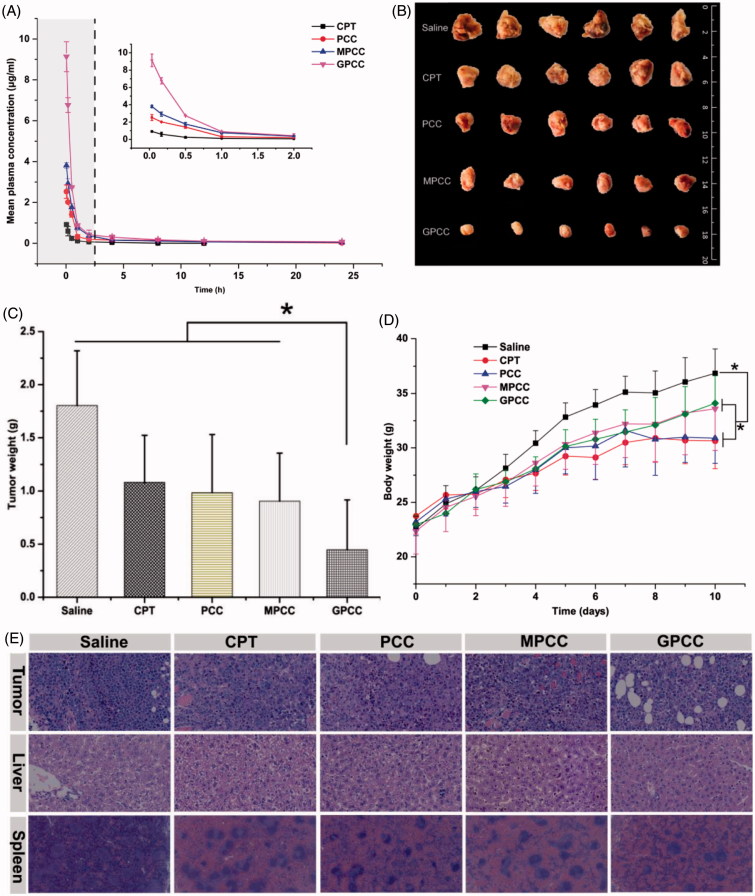
The efficient targeting of drugs to tumor cell and subsequent rapid drug release remain primary challenges in the development of nanomedicines for cancer therapy. Here, we constructed a glucose transporter 1 (GLUT1)-targeting and tumor cell microenvironment-sensitive drug release Glucose-PEG-PAMAM-s-s-Camptothecin-Cy7 (GPCC) conjugate to tackle the dilemma. The conjugate was characterized by a small particle size, spherical shape, and glutathione (GSH)-sensitive drug release. In vitro tumor targeting was explored in monolayer (2D) and multilayer tumor spheroid (3D) HepG2 cancer cell models (GLUT1+). The cellular uptake of GPCC was higher than that in the control groups and that in normal L02 cells (GLUT1-), likely due to the conjugated glucose moiety. Moreover, the GPCC conjugate exhibited stronger cytotoxicity, higher S arrest and enhanced apoptosis and necrosis rate in HepG2 cells than control groups but not L02 cells. However, the cytotoxicity of GPCC was lower than that of free CPT, which could be explained by the slower release of CPT from the GPCC compared with free CPT. Additional in vivo tumor targeting experiments demonstrated the superior tumor-targeting ability of the GPCC conjugate, which significantly accumulated in tumor meanwhile minimize in normal tissues compared with control groups. The GPCC conjugate showed better pharmacokinetic properties, enabling a prolonged circulation time and increased camptothecin area under the curve (AUC). These features contributed to better therapeutic efficacy and lower toxicity in H22 hepatocarcinoma tumor-bearing mice. The GLUT1-targeting, GSH-sensitive GPCC conjugate provides an efficient, safe and economic approach for tumor cell targeted drug delivery.
Keywords: GLUT1 targeting; PAMAM dendrimer; anticancer drug; conjugate; tumor microenvironment.
Conflict of interest statement
The authors report no declarations of interest.
Figures
References
-
- Barenholz YC. (2012). Doxil® – the first FDA-approved nano-drug: lessons learned. J Control Release 160:117–34. - PubMed
-
- Danhier F. (2016). To exploit the tumor microenvironment: since the EPR effect fails in the clinic, what is the future of nanomedicine. J Control Release 244:108–21. - PubMed
-
- Friedrich J, Seidel C, Ebner R, Kunz-Schughart LA. (2008). Spheroid-based drug screen: considerations and practical approach. Nature Protocols 4:309–24. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous