

Calmodulin fishing with a structurally disordered bait triggers CyaA catalysis
- PMID: 29287065
- PMCID: PMC5764468
- DOI: 10.1371/journal.pbio.2004486
Calmodulin fishing with a structurally disordered bait triggers CyaA catalysis
Abstract
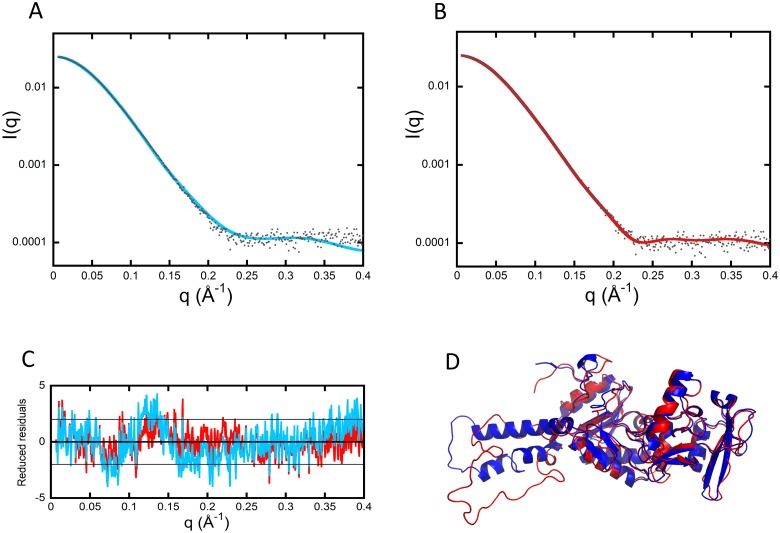
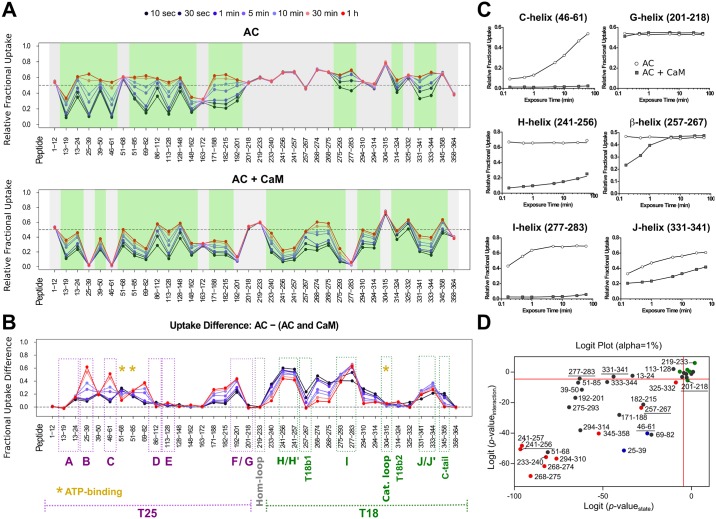
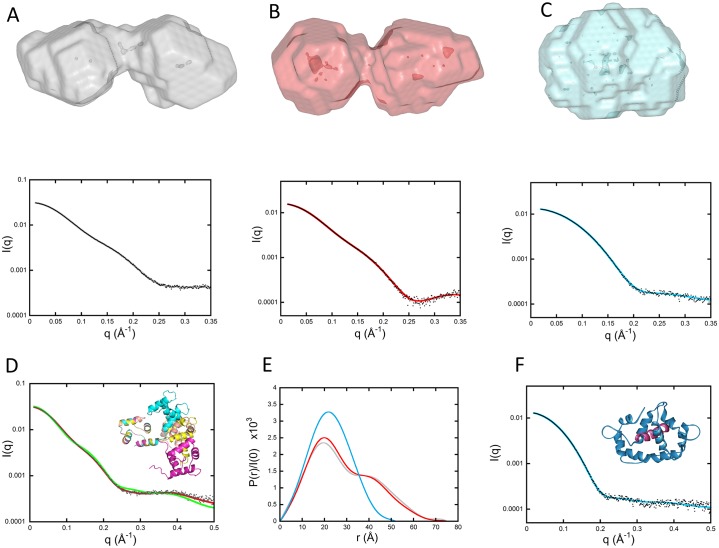
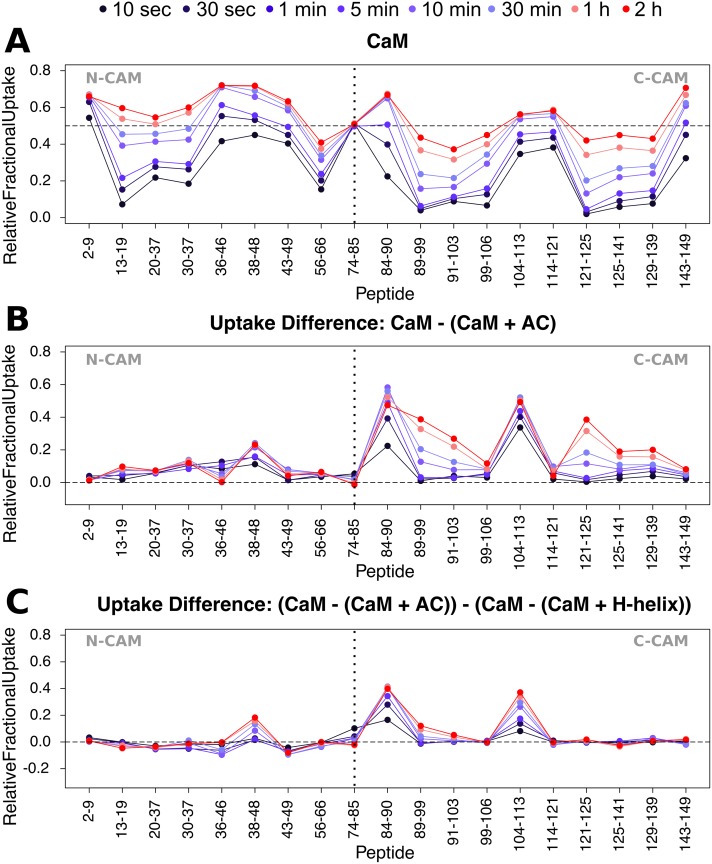
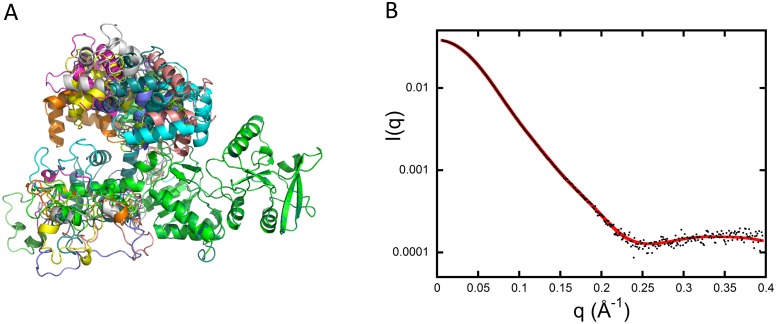
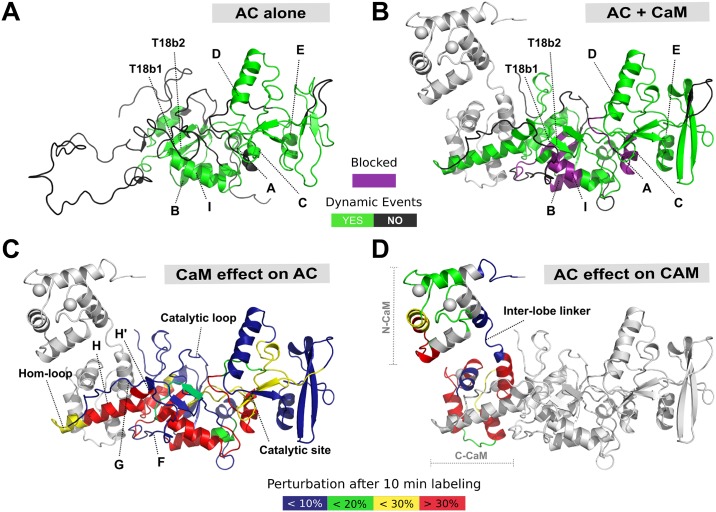
Once translocated into the cytosol of target cells, the catalytic domain (AC) of the adenylate cyclase toxin (CyaA), a major virulence factor of Bordetella pertussis, is potently activated by binding calmodulin (CaM) to produce supraphysiological levels of cAMP, inducing cell death. Using a combination of small-angle X-ray scattering (SAXS), hydrogen/deuterium exchange mass spectrometry (HDX-MS), and synchrotron radiation circular dichroism (SR-CD), we show that, in the absence of CaM, AC exhibits significant structural disorder, and a 75-residue-long stretch within AC undergoes a disorder-to-order transition upon CaM binding. Beyond this local folding, CaM binding induces long-range allosteric effects that stabilize the distant catalytic site, whilst preserving catalytic loop flexibility. We propose that the high enzymatic activity of AC is due to a tight balance between the CaM-induced decrease of structural flexibility around the catalytic site and the preservation of catalytic loop flexibility, allowing for fast substrate binding and product release. The CaM-induced dampening of AC conformational disorder is likely relevant to other CaM-activated enzymes.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Ladant D, Ullmann A. Bordetella pertussis adenylate cyclase: a toxin with multiple talents. Trends in microbiology. 1999;7(4):172–6. . - PubMed
-
- Tang WJ, Guo Q. The adenylyl cyclase activity of anthrax edema factor. Mol Aspects Med. 2009;30(6):423–30. doi: 10.1016/j.mam.2009.06.001 . - DOI - PMC - PubMed
-
- Karst JC, Barker R, Devi U, Swann MJ, Davi M, Roser SJ, et al. Identification of a region that assists membrane insertion and translocation of the catalytic domain of Bordetella pertussis CyaA toxin. J Biol Chem. 2012;287(12):9200–12. Epub 2012/01/14. doi: 10.1074/jbc.M111.316166 . - DOI - PMC - PubMed
-
- Cannella SE, Ntsogo Enguene VY, Davi M, Malosse C, Sotomayor Perez AC, Chamot-Rooke J, et al. Stability, structural and functional properties of a monomeric, calcium-loaded adenylate cyclase toxin, CyaA, from Bordetella pertussis. Sci Rep. 2017;7:42065 doi: 10.1038/srep42065 . - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
