

Mechanisms of Stress-induced Visceral Pain
- PMID: 29291604
- PMCID: PMC5753899
- DOI: 10.5056/jnm17137
Mechanisms of Stress-induced Visceral Pain
Abstract
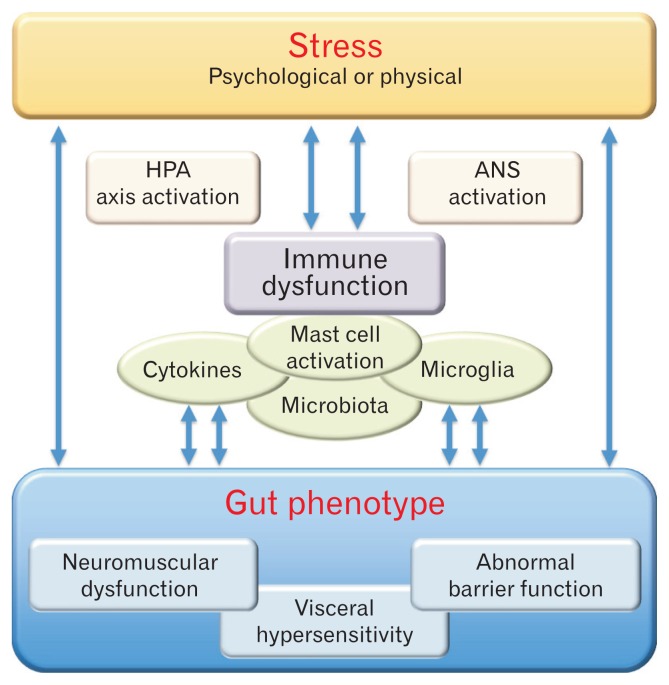
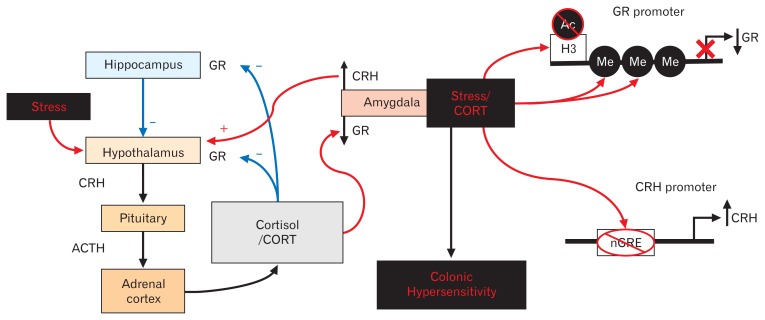
Evidence suggests that long-term stress facilitates visceral pain through sensitization of pain pathways and promotes chronic visceral pain disorders such as the irritable bowel syndrome (IBS). This review will describe the importance of stress in exacerbating IBS-induced abdominal pain. Additionally, we will briefly review our understanding of the activation of the hypothalamic-pituitary-adrenal axis by both chronic adult stress and following early life stress in the pathogenesis of IBS. The review will focus on the glucocorticoid receptor and corticotropin-releasing hormone-mediated mechanisms in the amygdala involved in stress-induced visceral hypersensitivity. One potential mechanism underlying persistent effects of stress on visceral sensitivity could be epigenetic modulation of gene expression. While there are relatively few studies examining epigenetically mediated mechanisms involved in stress-induced visceral nociception, alterations in DNA methylation and histone acetylation patterns within the brain, have been linked to alterations in nociceptive signaling via increased expression of pro-nociceptive neurotransmitters. This review will discuss the latest studies investigating the long-term effects of stress on visceral sensitivity. Additionally, we will critically review the importance of experimental models of adult stress and early life stress in enhancing our understanding of the basic molecular mechanisms of nociceptive processing.
Keywords: Amygdala; Early life adversity; Irritable bowel syndrome; Models; Visceral pain; animal.
Conflict of interest statement
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
