

Multi-trait analysis of genome-wide association summary statistics using MTAG
- PMID: 29292387
- PMCID: PMC5805593
- DOI: 10.1038/s41588-017-0009-4
Multi-trait analysis of genome-wide association summary statistics using MTAG
Erratum in
-
Publisher Correction: Multi-trait analysis of genome-wide association summary statistics using MTAG.Nat Genet. 2019 Jul;51(7):1190. doi: 10.1038/s41588-019-0444-5. Nat Genet. 2019. PMID: 31147634
-
Author Correction: Multi-trait analysis of genome-wide association summary statistics using MTAG.Nat Genet. 2019 Aug;51(8):1295. doi: 10.1038/s41588-019-0469-9. Nat Genet. 2019. PMID: 31239548
Abstract
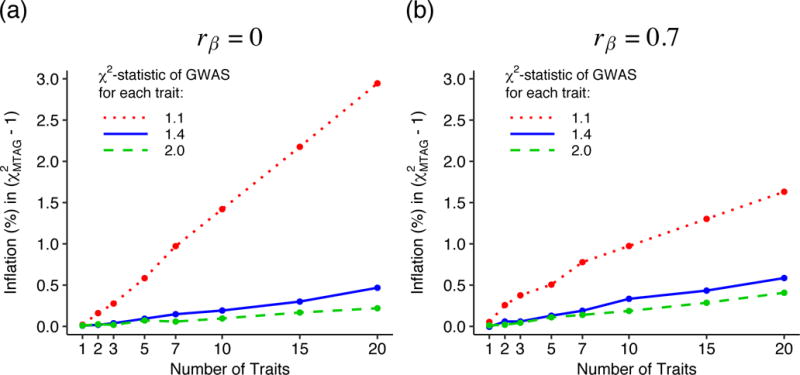
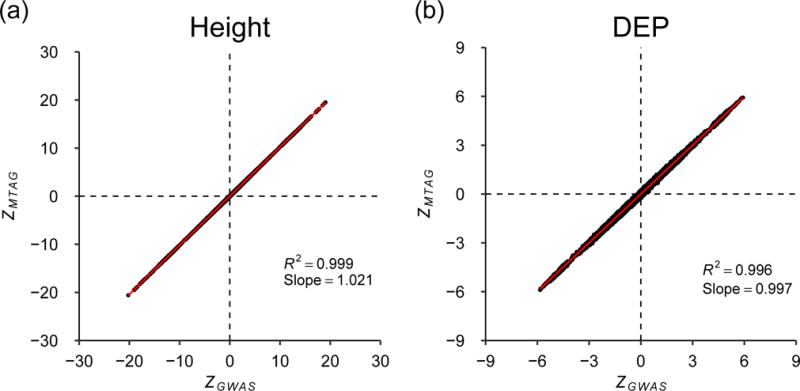
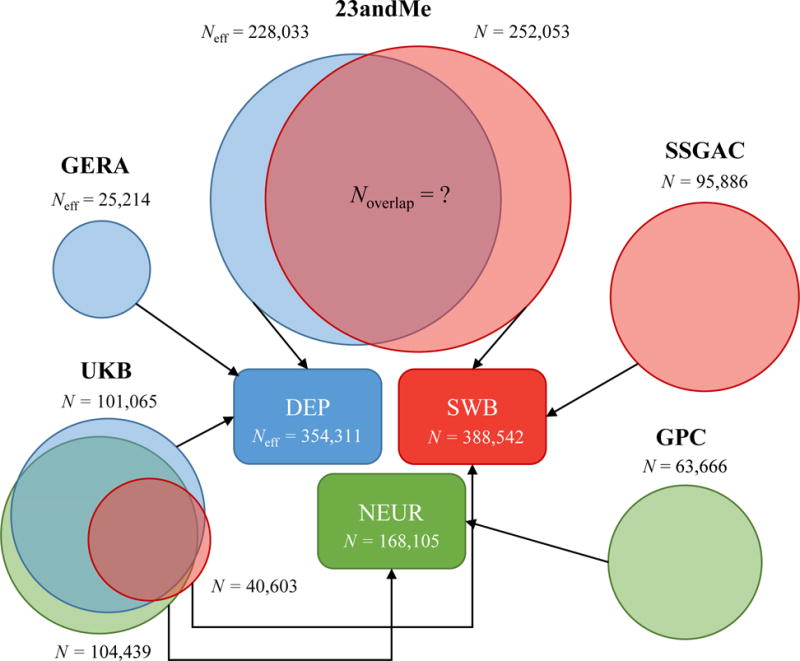
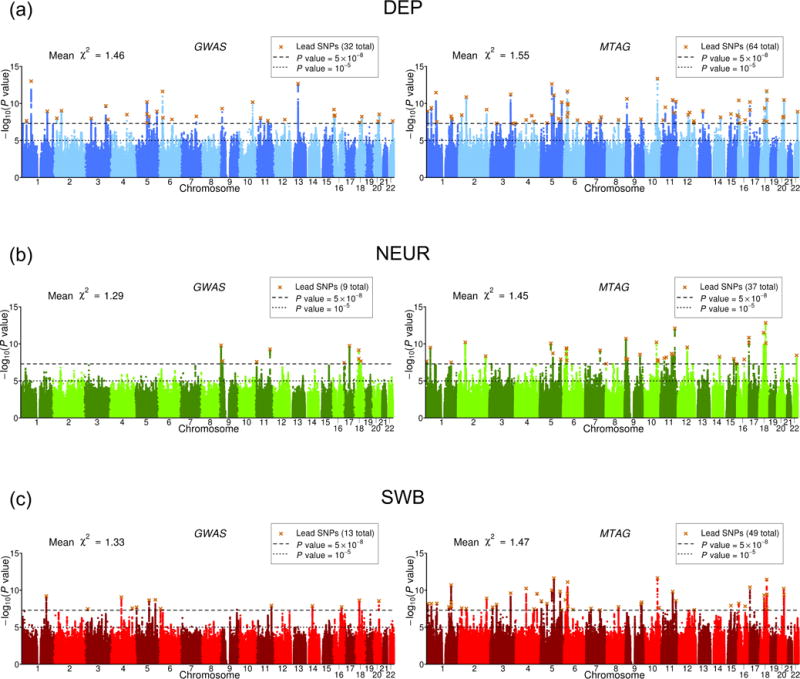
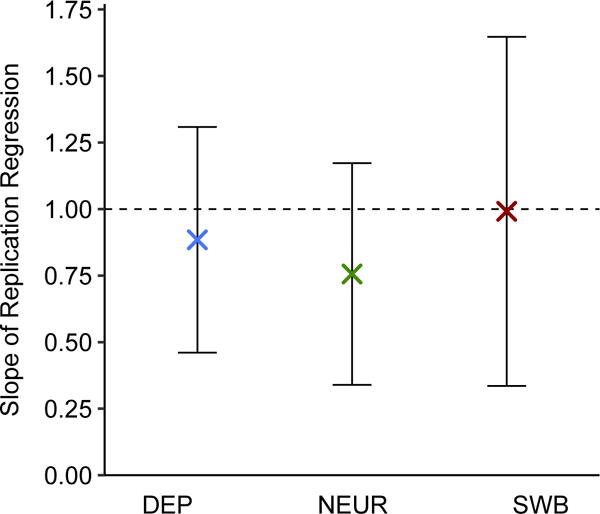
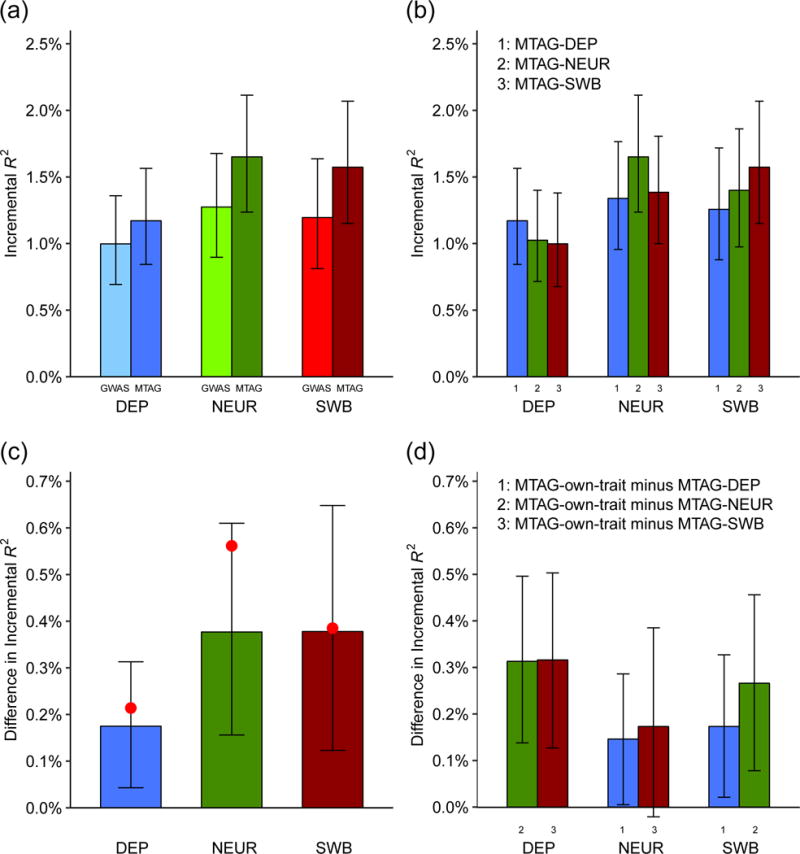
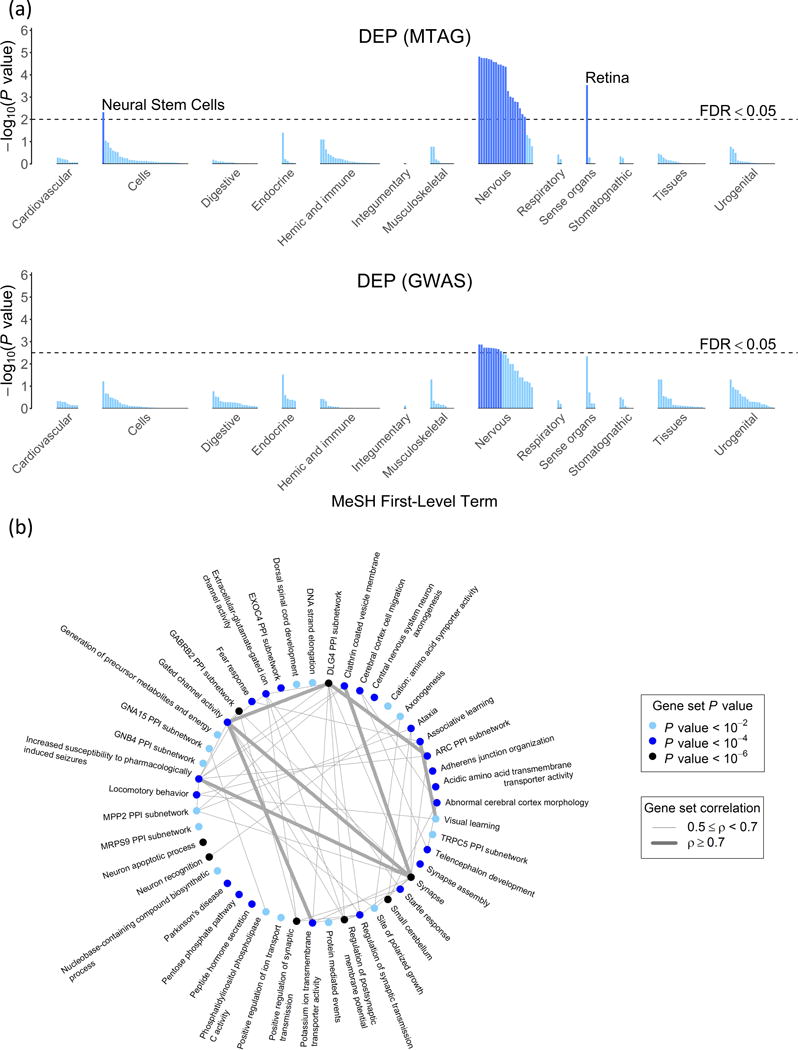
We introduce multi-trait analysis of GWAS (MTAG), a method for joint analysis of summary statistics from genome-wide association studies (GWAS) of different traits, possibly from overlapping samples. We apply MTAG to summary statistics for depressive symptoms (N eff = 354,862), neuroticism (N = 168,105), and subjective well-being (N = 388,538). As compared to the 32, 9, and 13 genome-wide significant loci identified in the single-trait GWAS (most of which are themselves novel), MTAG increases the number of associated loci to 64, 37, and 49, respectively. Moreover, association statistics from MTAG yield more informative bioinformatics analyses and increase the variance explained by polygenic scores by approximately 25%, matching theoretical expectations.
Conflict of interest statement
Figures
References
-
- Baselmans BML, et al. Multivariate Genome-Wide and Integrated Transcriptome and Epigenome-Wide Analyses of the Well-being Spectrum. bioRxiv. 2017 at < http://biorxiv.org/content/early/2017/03/11/115915.abstract>.
Publication types
MeSH terms
Grants and funding
- G1001799/MRC_/Medical Research Council/United Kingdom
- P01 HD031921/HD/NICHD NIH HHS/United States
- MR/N01104X/2/MRC_/Medical Research Council/United Kingdom
- R01 HD060726/HD/NICHD NIH HHS/United States
- MC_QA137853/MRC_/Medical Research Council/United Kingdom
- R01 HD073342/HD/NICHD NIH HHS/United States
- R01 MH107649/MH/NIMH NIH HHS/United States
- U01 MH109539/MH/NIMH NIH HHS/United States
- 647648/ERC_/European Research Council/International
- P30 AG034532/AG/NIA NIH HHS/United States
- R01 MH101244/MH/NIMH NIH HHS/United States
- P01 AG005842/AG/NIA NIH HHS/United States
- P30 AG012810/AG/NIA NIH HHS/United States
- R01 AG042568/AG/NIA NIH HHS/United States
- T32 AG000186/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
