

Improving pairwise comparison of protein sequences with domain co-occurrence
- PMID: 29293498
- PMCID: PMC5766236
- DOI: 10.1371/journal.pcbi.1005889
Improving pairwise comparison of protein sequences with domain co-occurrence
Abstract
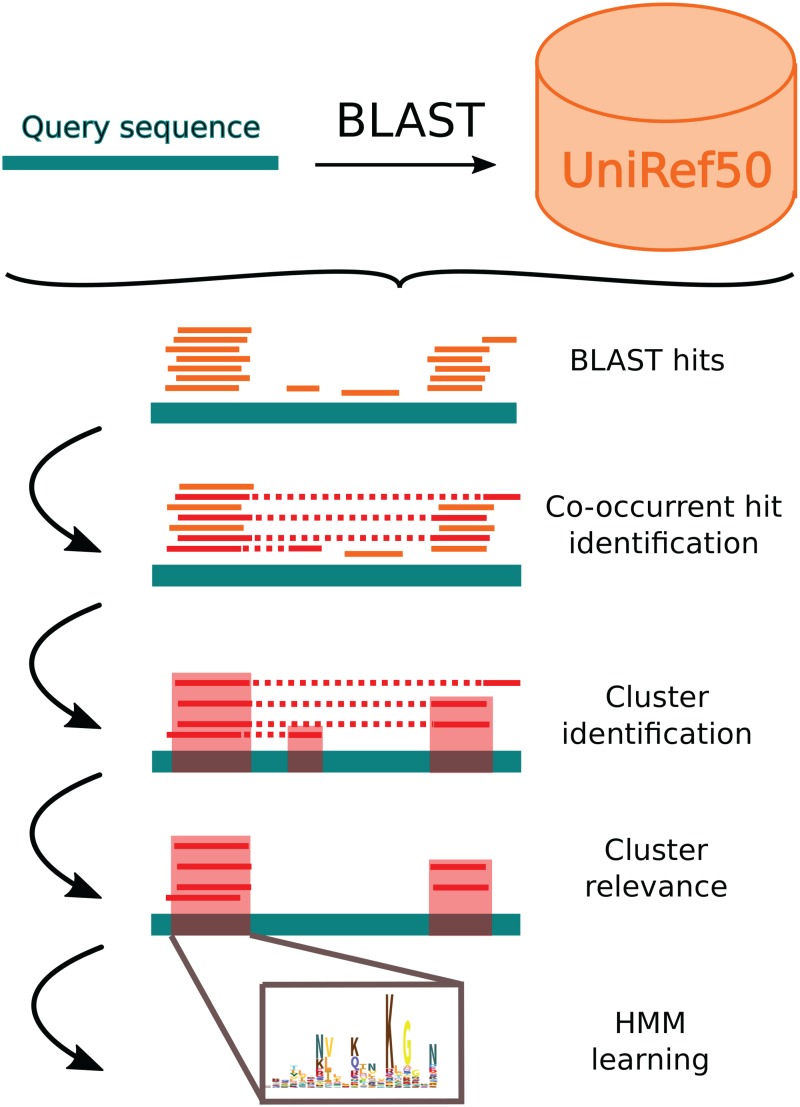
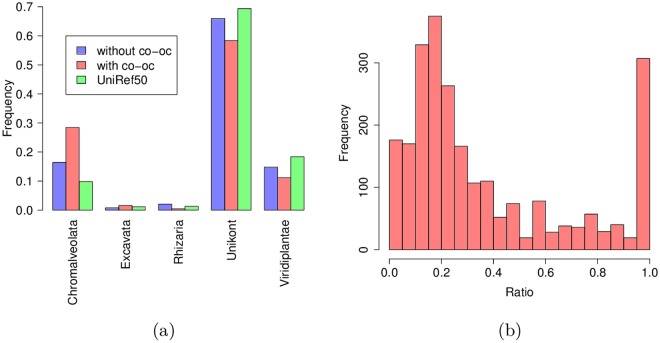
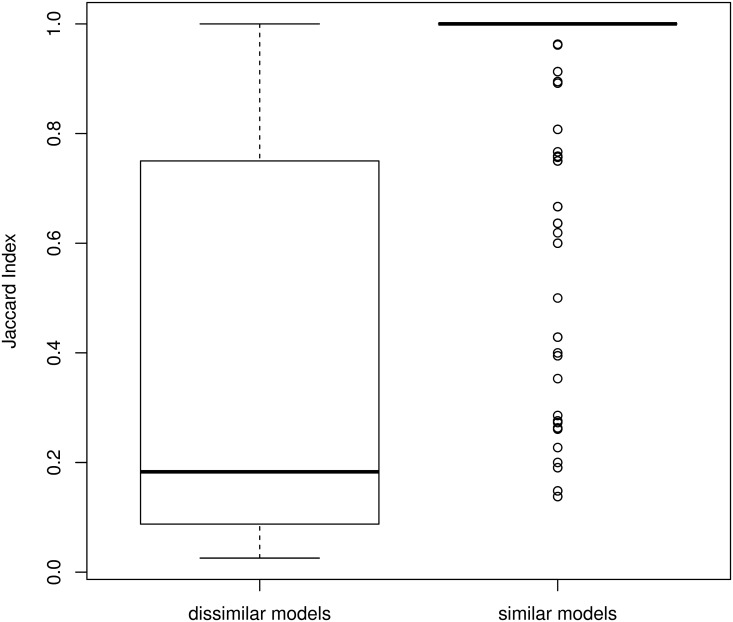
Comparing and aligning protein sequences is an essential task in bioinformatics. More specifically, local alignment tools like BLAST are widely used for identifying conserved protein sub-sequences, which likely correspond to protein domains or functional motifs. However, to limit the number of false positives, these tools are used with stringent sequence-similarity thresholds and hence can miss several hits, especially for species that are phylogenetically distant from reference organisms. A solution to this problem is then to integrate additional contextual information to the procedure. Here, we propose to use domain co-occurrence to increase the sensitivity of pairwise sequence comparisons. Domain co-occurrence is a strong feature of proteins, since most protein domains tend to appear with a limited number of other domains on the same protein. We propose a method to take this information into account in a typical BLAST analysis and to construct new domain families on the basis of these results. We used Plasmodium falciparum as a case study to evaluate our method. The experimental findings showed an increase of 14% of the number of significant BLAST hits and an increase of 25% of the proteome area that can be covered with a domain. Our method identified 2240 new domains for which, in most cases, no model of the Pfam database could be linked. Moreover, our study of the quality of the new domains in terms of alignment and physicochemical properties show that they are close to that of standard Pfam domains. Source code of the proposed approach and supplementary data are available at: https://gite.lirmm.fr/menichelli/pairwise-comparison-with-cooccurrence.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures









References
-
- Zmasek CM, Godzik A. Strong functional patterns in the evolution of eukaryotic genomes revealed by the reconstruction of ancestral protein domain repertoires. Genome Biology. 2011;12(1):R4 doi: 10.1186/gb-2011-12-1-r4 - DOI - PMC - PubMed
-
- Bornberg-Bauer E, Albà MM. Dynamics and adaptive benefits of modular protein evolution. Current Opinion in Structural Biology. 2013;23(3):459–466. doi: 10.1016/j.sbi.2013.02.012 - DOI - PubMed
-
- Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Research. 2016;44(D1):D279–D285. doi: 10.1093/nar/gkv1344 - DOI - PMC - PubMed
-
- Durbin R, Eddy S, Krogh A, Mitchison G. Biological sequence analysis probabilistic models of proteins and nucleic acids. Cambridge University Press; 1998.
-
- Terrapon N, Gascuel O, Maréchal E, Bréhélin L. Detection of new protein domains using co-occurrence: application to Plasmodium falciparum. Bioinformatics. 2009;25(23):3077–3083. doi: 10.1093/bioinformatics/btp560 - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
