

XPAT: a toolkit to conduct cross-platform association studies with heterogeneous sequencing datasets
- PMID: 29294048
- PMCID: PMC5888834
- DOI: 10.1093/nar/gkx1280
XPAT: a toolkit to conduct cross-platform association studies with heterogeneous sequencing datasets
Abstract
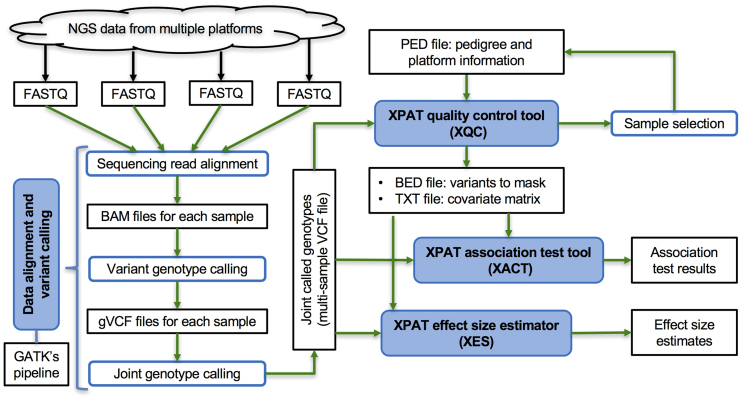
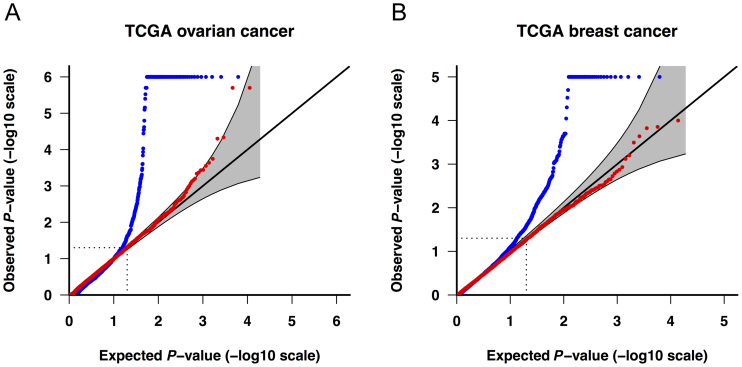
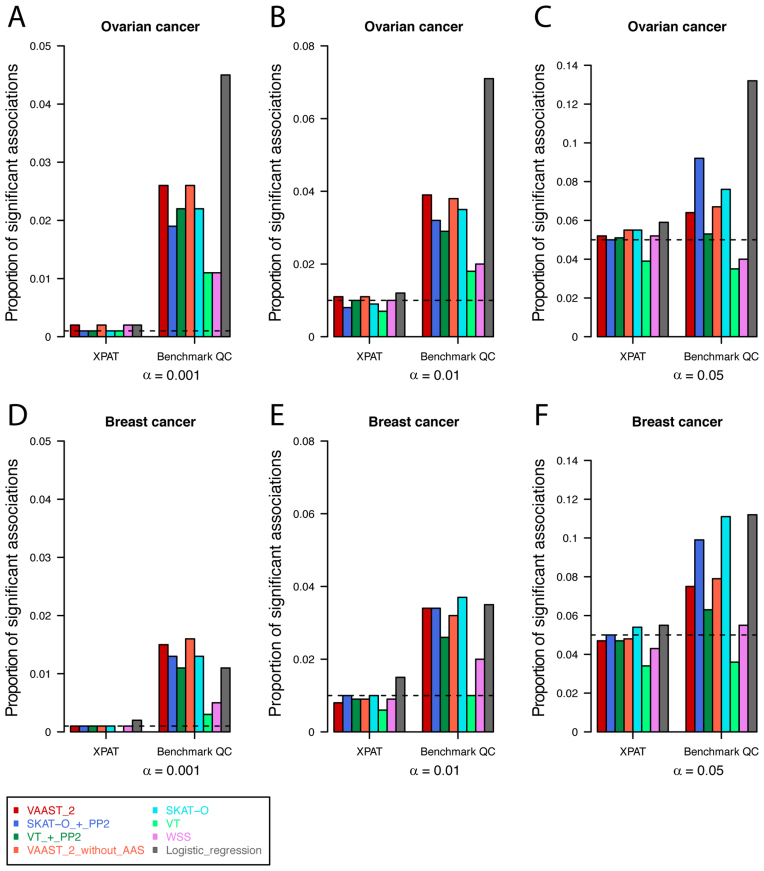
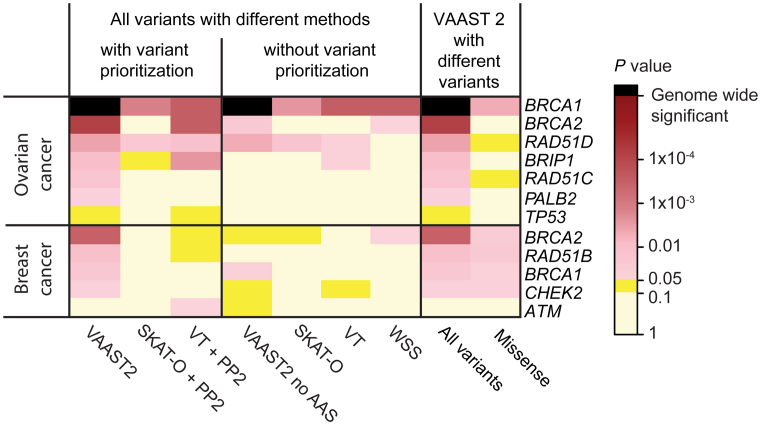
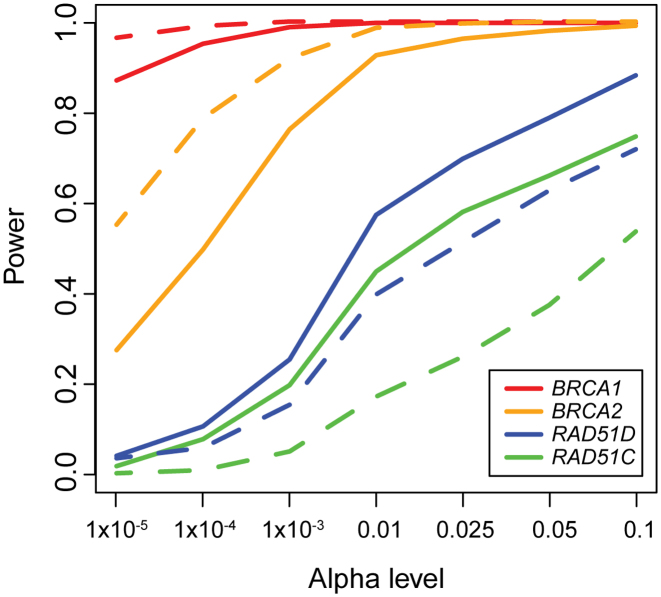
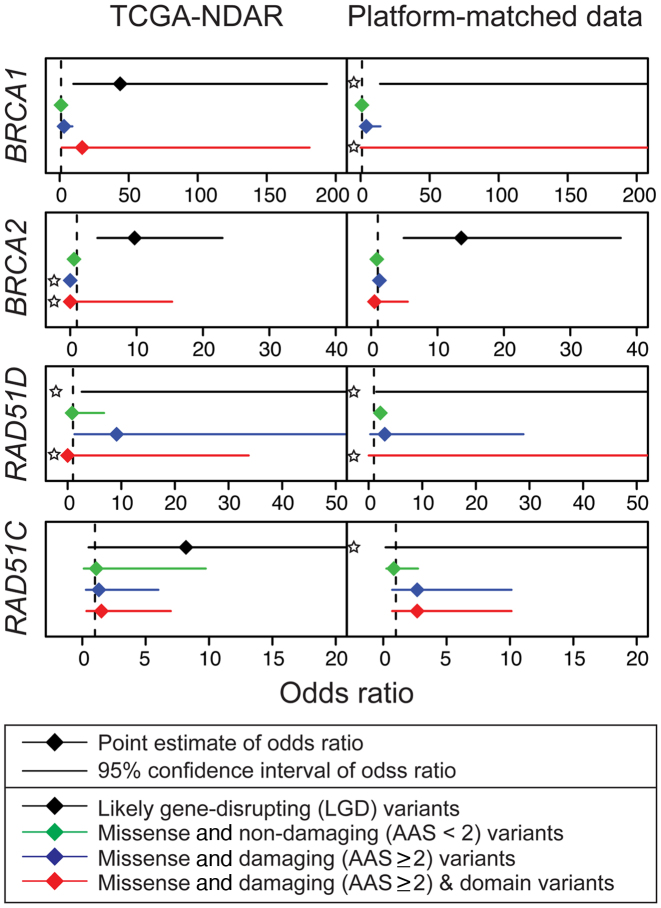
High-throughput sequencing data are increasingly being made available to the research community for secondary analyses, providing new opportunities for large-scale association studies. However, heterogeneity in target capture and sequencing technologies often introduce strong technological stratification biases that overwhelm subtle signals of association in studies of complex traits. Here, we introduce the Cross-Platform Association Toolkit, XPAT, which provides a suite of tools designed to support and conduct large-scale association studies with heterogeneous sequencing datasets. XPAT includes tools to support cross-platform aware variant calling, quality control filtering, gene-based association testing and rare variant effect size estimation. To evaluate the performance of XPAT, we conducted case-control association studies for three diseases, including 783 breast cancer cases, 272 ovarian cancer cases, 205 Crohn disease cases and 3507 shared controls (including 1722 females) using sequencing data from multiple sources. XPAT greatly reduced Type I error inflation in the case-control analyses, while replicating many previously identified disease-gene associations. We also show that association tests conducted with XPAT using cross-platform data have comparable performance to tests using matched platform data. XPAT enables new association studies that combine existing sequencing datasets to identify genetic loci associated with common diseases and other complex traits.
Figures
References
-
- Evangelou E., Ioannidis J.P.. Meta-analysis methods for genome-wide association studies and beyond. Nat. Rev. Genet. 2013; 14:379–389. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
