

Immunomodulatory, liver depot gene therapy for Pompe disease
- PMID: 29295737
- PMCID: PMC6026080
- DOI: 10.1016/j.cellimm.2017.12.011
Immunomodulatory, liver depot gene therapy for Pompe disease
Abstract
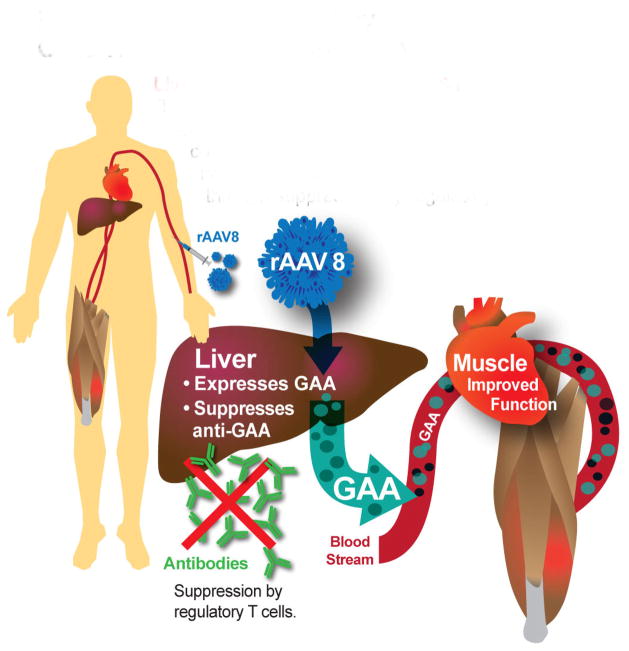
Pompe disease is caused by mutations in acid alpha glucosidase (GAA) that causes accumulation of lysosomal glycogen affecting the heart and skeletal muscles, and can be fatal. Enzyme replacement therapy (ERT) with recombinant human GAA (rhGAA) improves muscle function by reducing glycogen accumulation. Limitations of ERT include a short half-life and the formation of antibodies that result in reduced efficacy. By harnessing the immune tolerance induction properties of the liver, liver-targeted gene delivery (with an adeno-associated virus vector containing a liver specific promoter), suppresses immunity against the GAA introduced by gene therapy. This induces immune tolerance to rhGAA by activating regulatory T cells and simultaneously, corrects GAA deficiency. Potentially, liver-targeted gene therapy can be performed once with lasting effects, by administering a relatively low dose of an adeno-associated virus type 8 vector to replace and induce immune tolerance to GAA.
Keywords: Acid alpha-glucosidase; Antibody response; Enzyme replacement therapy; Gene therapy; Glycogen storage disease; Immune tolerance; Pompe disease.
Copyright © 2018 Elsevier Inc. All rights reserved.
Conflict of interest statement
Figures
References
-
- Hopkins PV, Campbell C, Klug T, et al. Lysosomal storage disorder screening implementation: findings from the first six months of full population pilot testing in Missouri. J Pediatr. 2015;166:172–177. - PubMed
-
- Kishnani PS, Hwu WL, Mandel H, et al. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr. 2006;148:671–676. - PubMed
-
- Case LE, Bjartmar C, Morgan C, et al. Safety and efficacy of alternative alglucosidase alfa regimens in Pompe disease. Neuromuscul Disord. 2015;25:321–332. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
