

Atypical periodic paralysis and myalgia: A novel RYR1 phenotype
- PMID: 29298851
- PMCID: PMC5791790
- DOI: 10.1212/WNL.0000000000004894
Atypical periodic paralysis and myalgia: A novel RYR1 phenotype
Abstract
Objective: To characterize the phenotype of patients with symptoms of periodic paralysis (PP) and ryanodine receptor (RYR1) gene mutations.
Methods: Cases with a possible diagnosis of PP but additional clinicopathologic findings previously associated with RYR1-related disorders were referred for a tertiary neuromuscular clinical assessment in which they underwent detailed clinical evaluation, including neurophysiologic assessment, muscle biopsy, and muscle MRI. Genetic analysis with next-generation sequencing and/or targeted Sanger sequencing was performed.
Results: Three cases with episodic muscle paralysis or weakness and additional findings compatible with a RYR1-related myopathy were identified. The McManis test, used in the diagnosis of PP, was positive in 2 of 3 cases. Genetic analysis of known PP genes was negative. RYR1 analysis confirmed likely pathogenic variants in all 3 cases.
Conclusions: RYR1 mutations can cause late-onset atypical PP both with and without associated myopathy. Myalgia and cramps are prominent features. The McManis test may be a useful diagnostic tool to indicate RYR1-associated PP. We propose that clinicopathologic features suggestive of RYR1-related disorders should be sought in genetically undefined PP cases and that RYR1 gene testing be considered in those in whom mutations in SCN4A, CACNA1S, and KCNJ2 have already been excluded.
© 2018 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology.
Figures
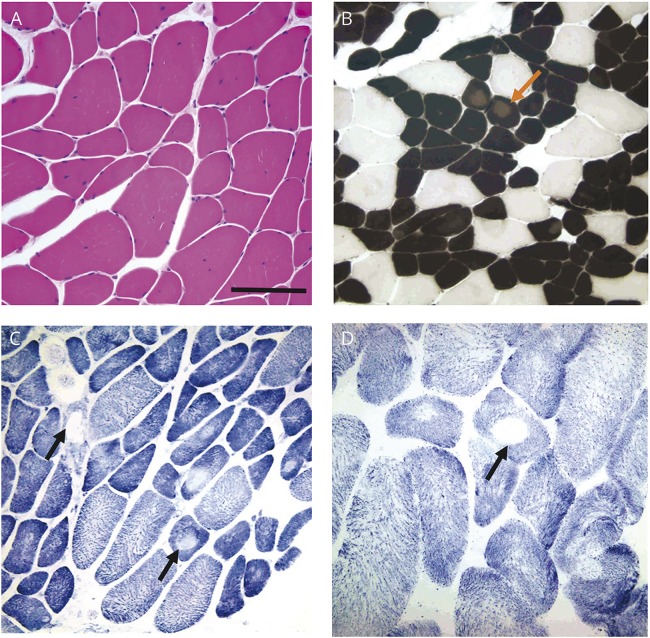
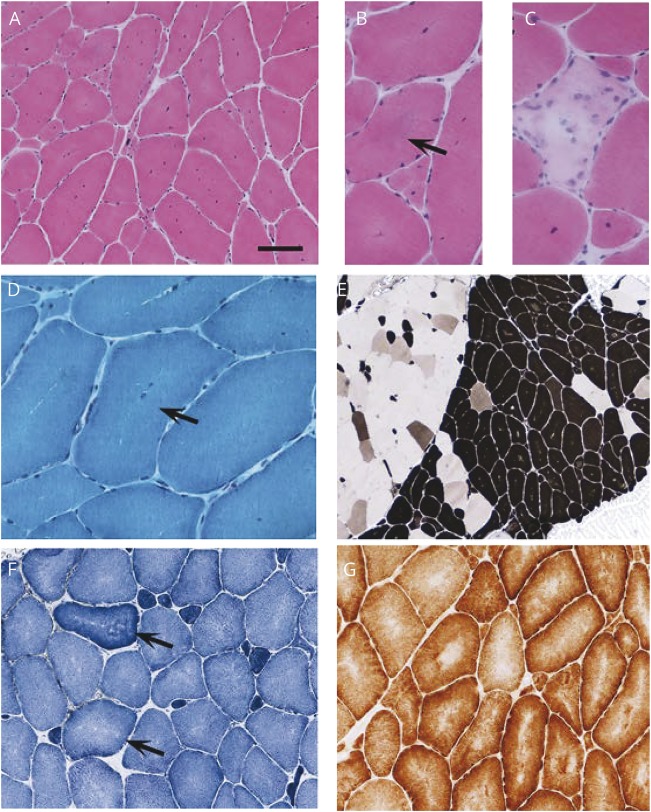
References
-
- Amburgey K, McNamara N, Bennett LR, McCormick ME, Acsadi G, Dowling JJ. Prevalence of congenital myopathies in a representative pediatric United States population. Ann Neurol 2011;70:662–665. - PubMed
-
- Maggi L, Scoto M, Cirak S, et al. . Congenital myopathies: clinical features and frequency of individual subtypes diagnosed over a 5-year period in the United Kingdom. Neuromuscul Disord 2013;23:195–205. - PubMed
-
- Zhang Y, Chen HS, Khanna VK, et al. . A mutation in the human ryanodine receptor gene associated with central core disease. Nat Genet 1993;5:46–50. - PubMed
-
- Jungbluth H, Zhou H, Hartley L, et al. . Minicore myopathy with ophthalmoplegia caused by mutations in the ryanodine receptor type 1 gene. Neurology 2005;65:1930–1935. - PubMed
-
- Wilmshurst JM, Lillis S, Zhou H, et al. . RYR1 mutations are a common cause of congenital myopathies with central nuclei. Ann Neurol 2010;68:717–726. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous