

Class 1-Selective Histone Deacetylase (HDAC) Inhibitors Enhance HIV Latency Reversal while Preserving the Activity of HDAC Isoforms Necessary for Maximal HIV Gene Expression
- PMID: 29298886
- PMCID: PMC5827401
- DOI: 10.1128/JVI.02110-17
Class 1-Selective Histone Deacetylase (HDAC) Inhibitors Enhance HIV Latency Reversal while Preserving the Activity of HDAC Isoforms Necessary for Maximal HIV Gene Expression
Abstract
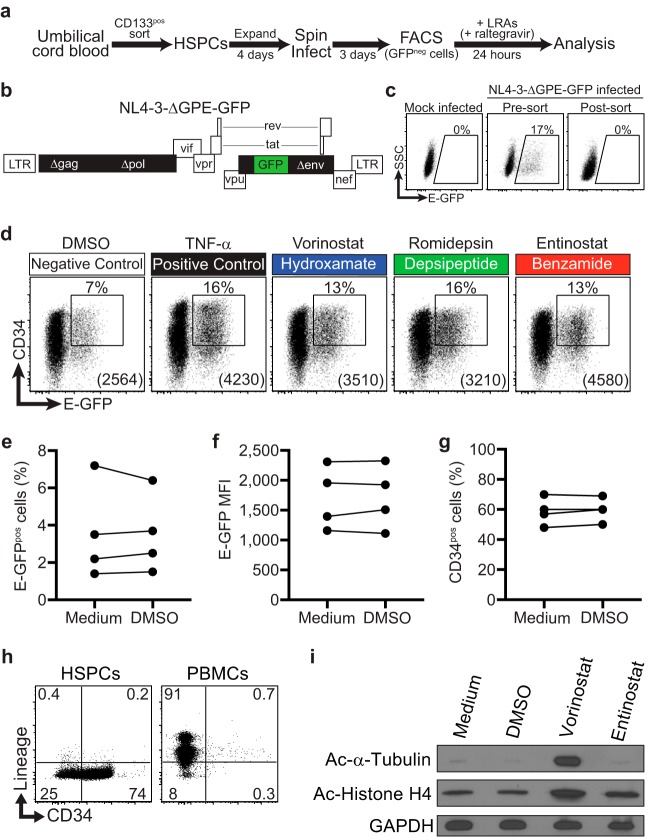
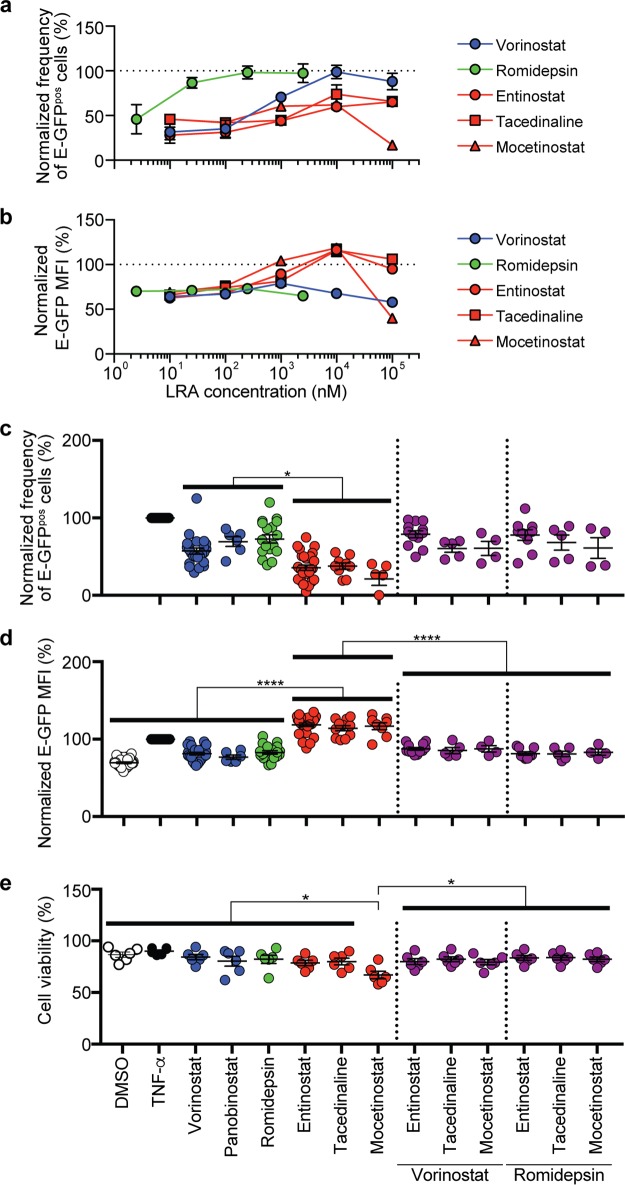
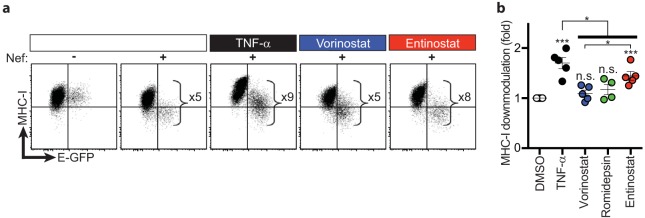
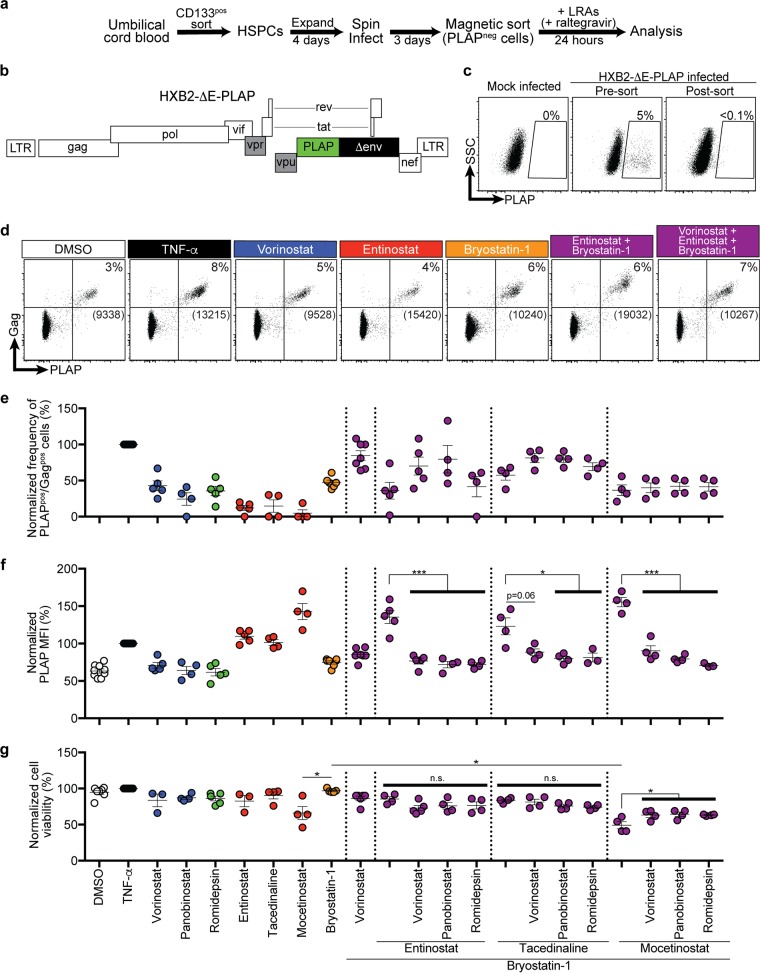
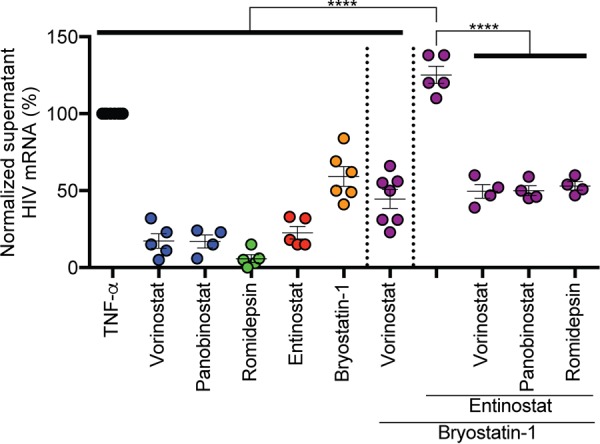
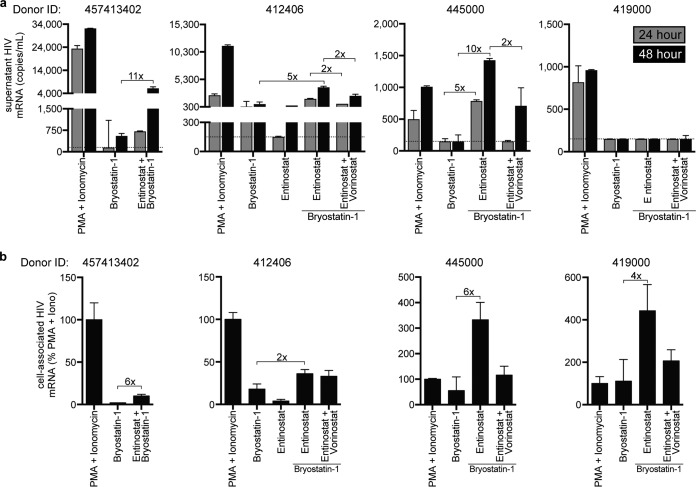
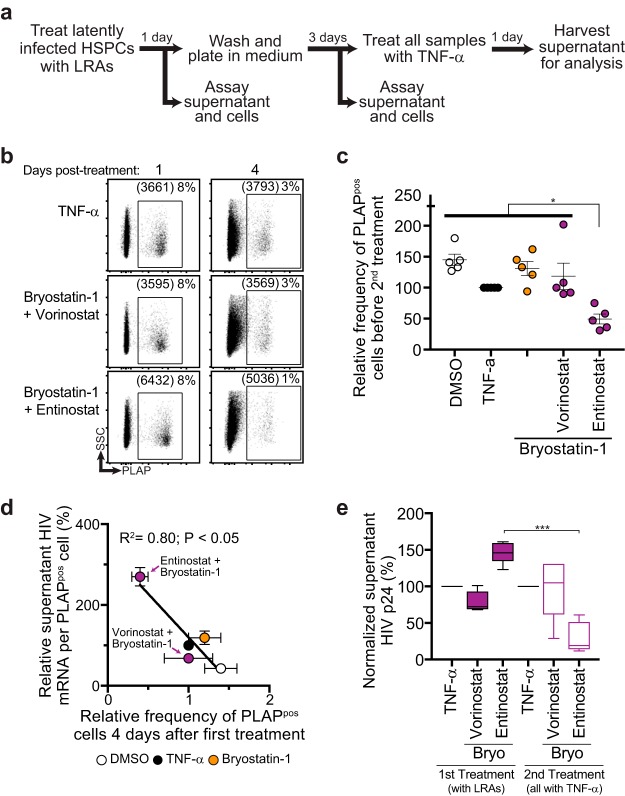
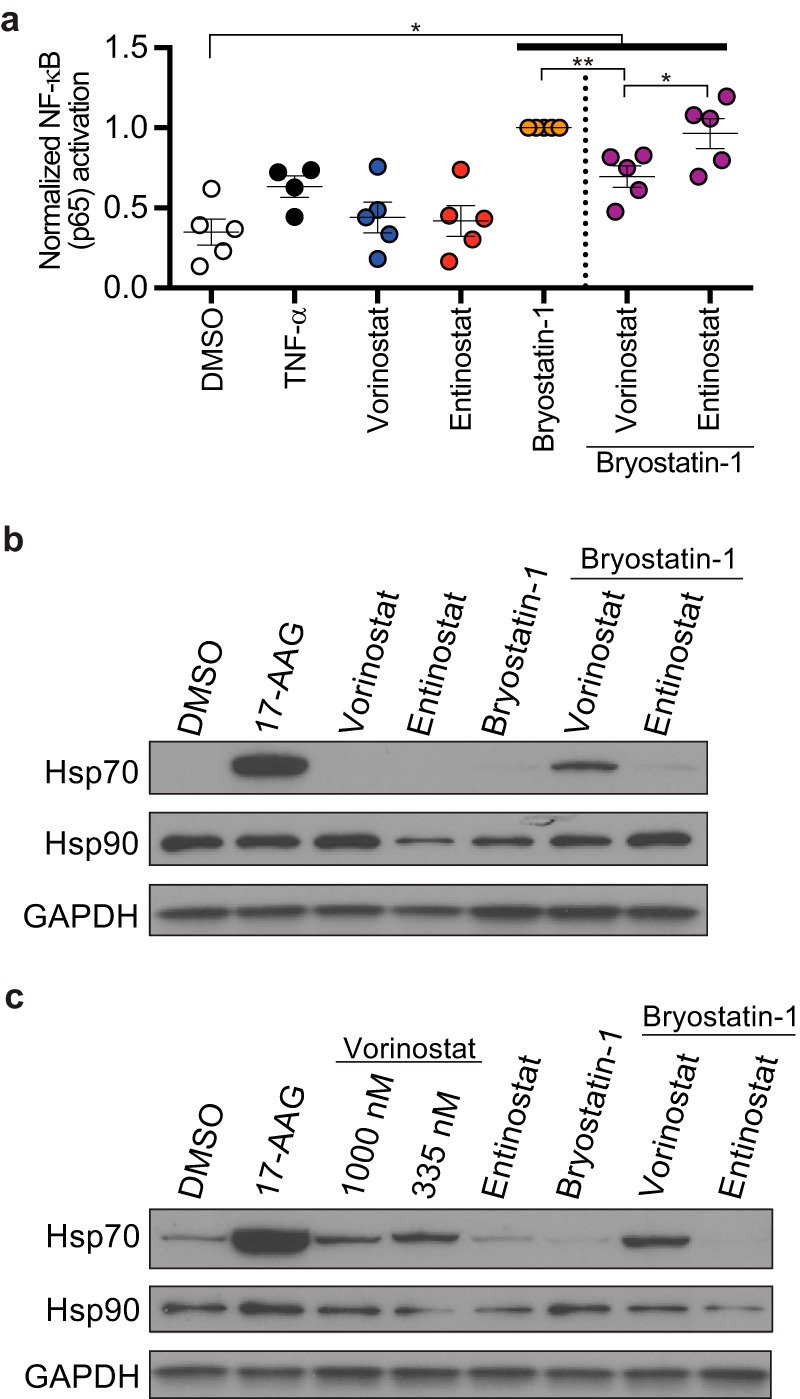
Combinations of drugs that affect distinct mechanisms of HIV latency aim to induce robust latency reversal leading to cytopathicity and elimination of the persistent HIV reservoir. Thus far, attempts have focused on combinations of protein kinase C (PKC) agonists and pan-histone deacetylase inhibitors (HDIs) despite the knowledge that HIV gene expression is regulated by class 1 histone deacetylases. We hypothesized that class 1-selective HDIs would promote more robust HIV latency reversal in combination with a PKC agonist than pan-HDIs because they preserve the activity of proviral factors regulated by non-class 1 histone deacetylases. Here, we show that class 1-selective agents used alone or with the PKC agonist bryostatin-1 induced more HIV protein expression per infected cell. In addition, the combination of entinostat and bryostatin-1 induced viral outgrowth, whereas bryostatin-1 combinations with pan-HDIs did not. When class 1-selective HDIs were used in combination with pan-HDIs, the amount of viral protein expression and virus outgrowth resembled that of pan-HDIs alone, suggesting that pan-HDIs inhibit robust gene expression induced by class 1-selective HDIs. Consistent with this, pan-HDI-containing combinations reduced the activity of NF-κB and Hsp90, two cellular factors necessary for potent HIV protein expression, but did not significantly reduce overall cell viability. An assessment of viral clearance from in vitro cultures indicated that maximal protein expression induced by class 1-selective HDI treatment was crucial for reservoir clearance. These findings elucidate the limitations of current approaches and provide a path toward more effective strategies to eliminate the HIV reservoir.IMPORTANCE Despite effective antiretroviral therapy, HIV evades eradication in a latent form that is not affected by currently available drug regimens. Pharmacologic latency reversal that leads to death of cellular reservoirs has been proposed as a strategy for reservoir elimination. Because histone deacetylases (HDACs) promote HIV latency, HDAC inhibitors have been a focus of HIV cure research. However, many of these inhibitors broadly affect multiple classes of HDACs, including those that promote HIV gene expression (class 1 HDACs). Here, we demonstrate that targeted treatment with class 1-selective HDAC inhibitors induced more potent HIV latency reversal than broadly acting agents. Additionally, we provide evidence that broadly acting HDIs are limited by inhibitory effects on non-class 1 HDACs that support the activity of proviral factors. Thus, our work demonstrates that the use of targeted approaches to induce maximum latency reversal affords the greatest likelihood of reservoir elimination.
Keywords: HIV; Hsp90; histone deacetylase inhibitors; latency reversal; shock and kill.
Copyright © 2018 American Society for Microbiology.
Figures
References
-
- Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, Quinn TC, Chadwick K, Margolick J, Brookmeyer R, Gallant J, Markowitz M, Ho DD, Richman DD, Siliciano RF. 1997. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278:1295–1300. doi: 10.1126/science.278.5341.1295. - DOI - PubMed
-
- Chun TW, Carruth L, Finzi D, Shen X, DiGiuseppe JA, Taylor H, Hermankova M, Chadwick K, Margolick J, Quinn TC, Kuo YH, Brookmeyer R, Zeiger MA, Barditch-Crovo P, Siliciano RF. 1997. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 387:183–188. doi: 10.1038/387183a0. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
