

A machine learning approach to integrate big data for precision medicine in acute myeloid leukemia
- PMID: 29298978
- PMCID: PMC5752671
- DOI: 10.1038/s41467-017-02465-5
A machine learning approach to integrate big data for precision medicine in acute myeloid leukemia
Abstract
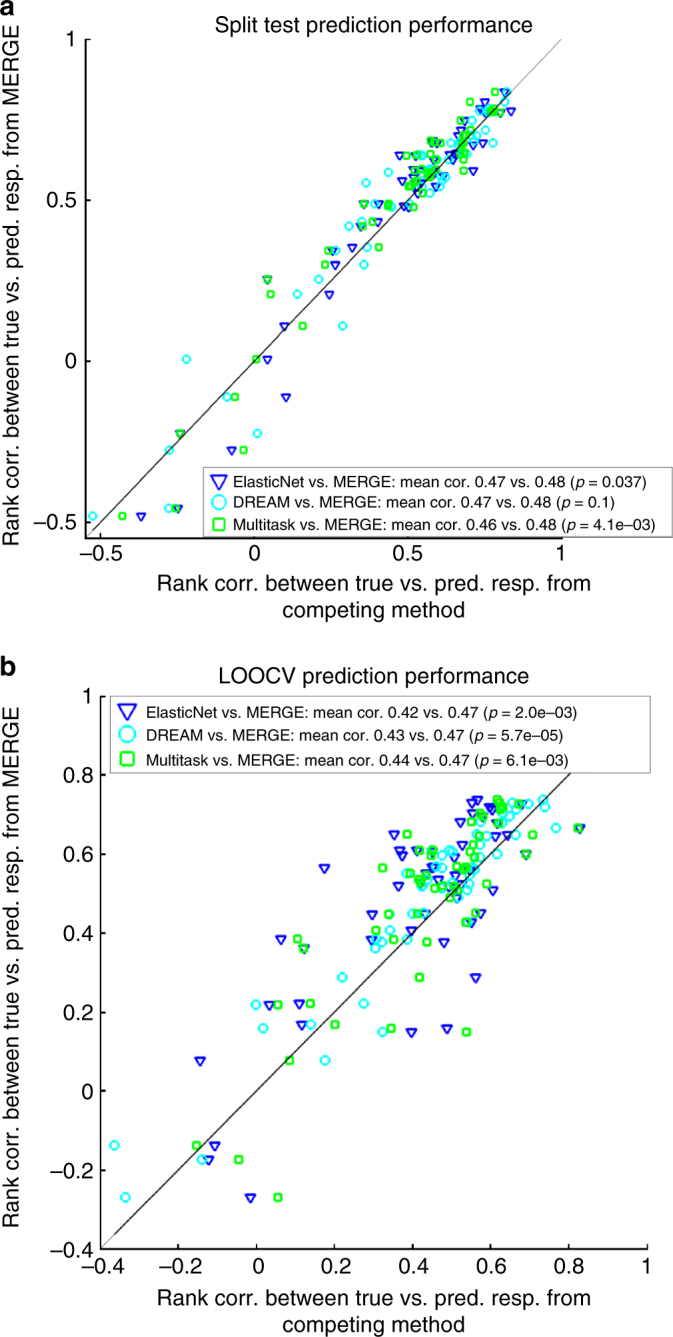
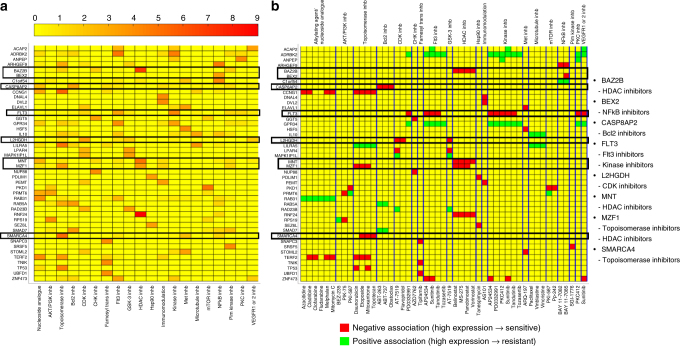
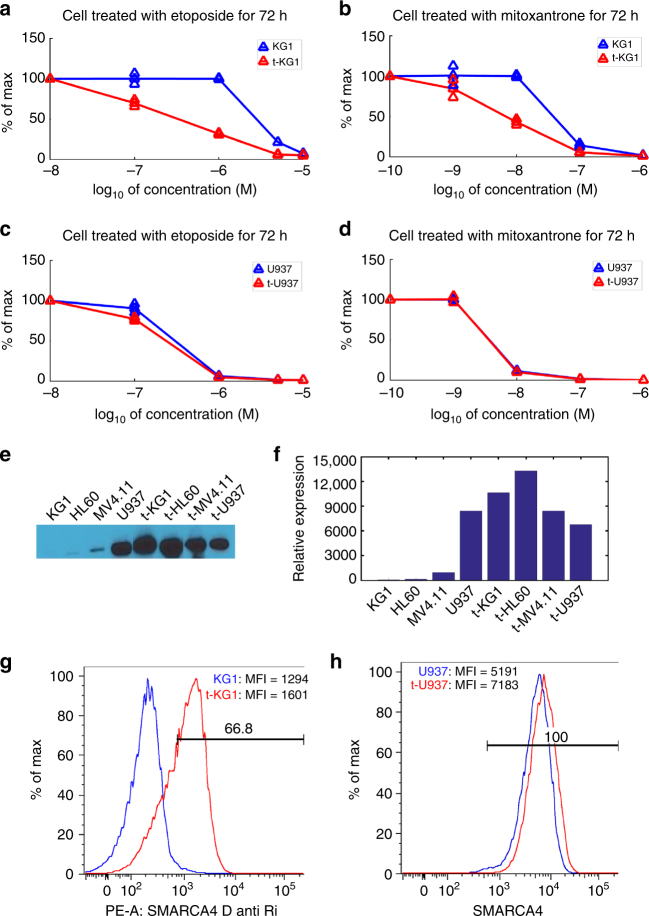
Cancers that appear pathologically similar often respond differently to the same drug regimens. Methods to better match patients to drugs are in high demand. We demonstrate a promising approach to identify robust molecular markers for targeted treatment of acute myeloid leukemia (AML) by introducing: data from 30 AML patients including genome-wide gene expression profiles and in vitro sensitivity to 160 chemotherapy drugs, a computational method to identify reliable gene expression markers for drug sensitivity by incorporating multi-omic prior information relevant to each gene's potential to drive cancer. We show that our method outperforms several state-of-the-art approaches in identifying molecular markers replicated in validation data and predicting drug sensitivity accurately. Finally, we identify SMARCA4 as a marker and driver of sensitivity to topoisomerase II inhibitors, mitoxantrone, and etoposide, in AML by showing that cell lines transduced to have high SMARCA4 expression reveal dramatically increased sensitivity to these agents.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
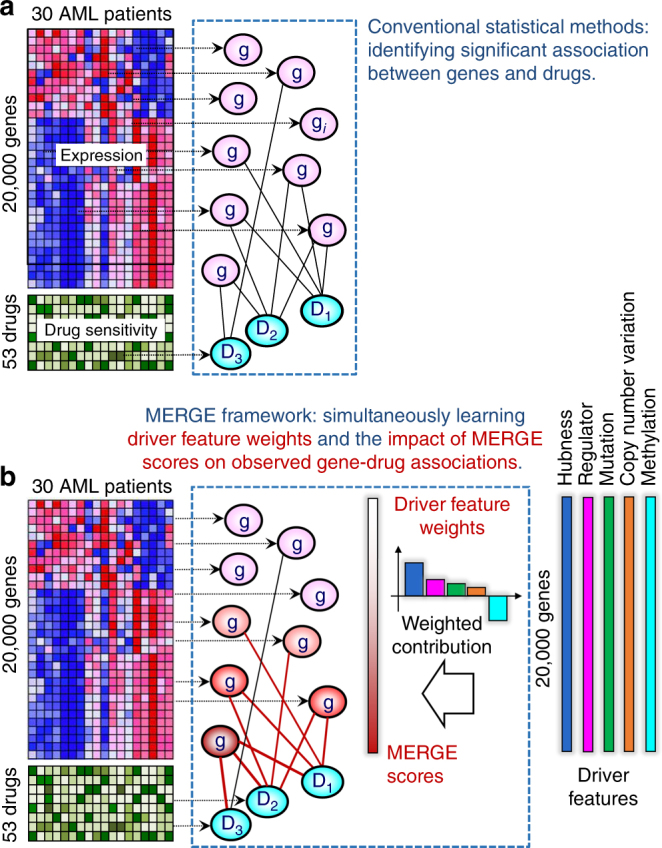
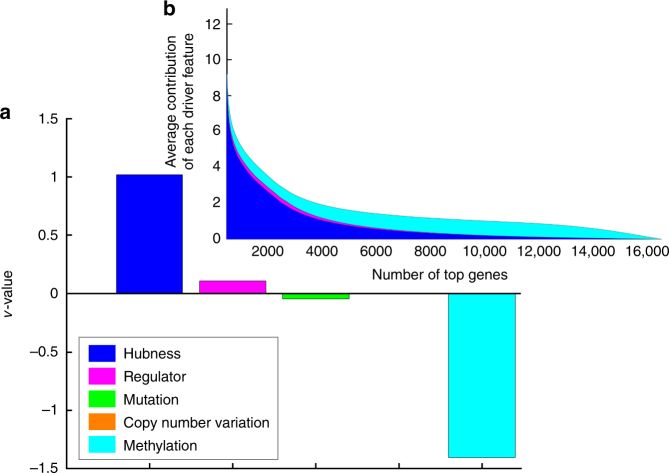
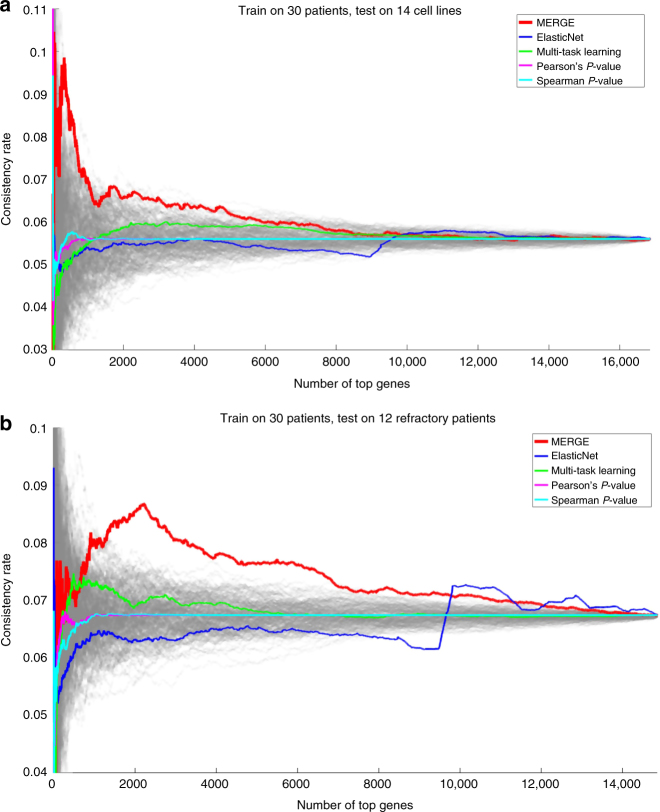
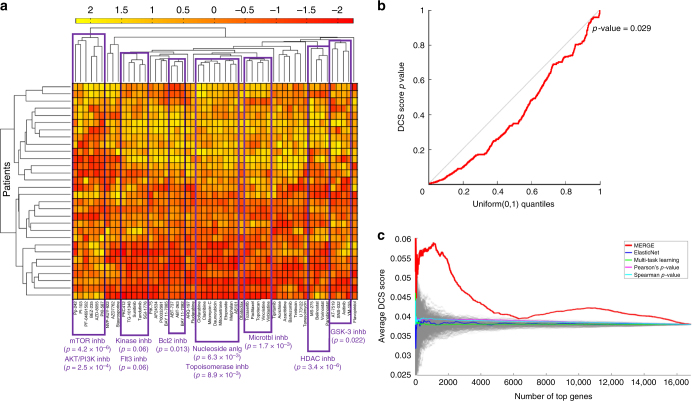
References
-
- PhRMA. Summer 2016 chart pack of the Pharmaceutical Research and Manufacturers of America (PhRMA, 2016).
-
- Zou H, Hastie T. Regularization and variable selection via the elastic net. J. R. Stat. Soc. Ser. B. 2005;67:768–768. doi: 10.1111/j.1467-9868.2005.00527.x. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
