

Construction of the third-generation Zea mays haplotype map
- PMID: 29300887
- PMCID: PMC5890452
- DOI: 10.1093/gigascience/gix134
Construction of the third-generation Zea mays haplotype map
Abstract
Background: Characterization of genetic variations in maize has been challenging, mainly due to deterioration of collinearity between individual genomes in the species. An international consortium of maize research groups combined resources to develop the maize haplotype version 3 (HapMap 3), built from whole-genome sequencing data from 1218 maize lines, covering predomestication and domesticated Zea mays varieties across the world.
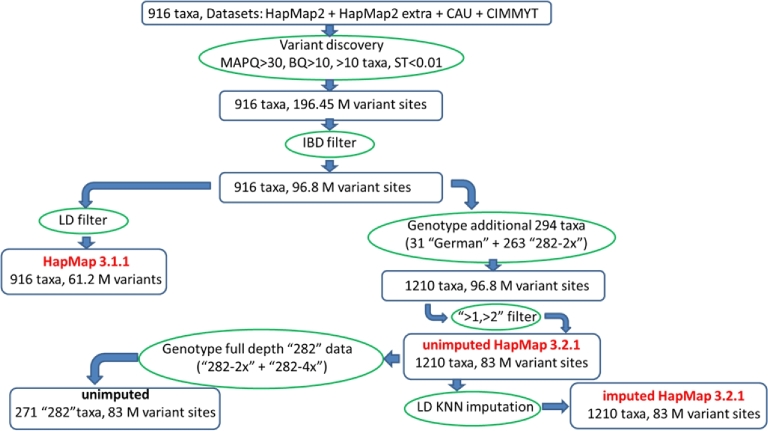
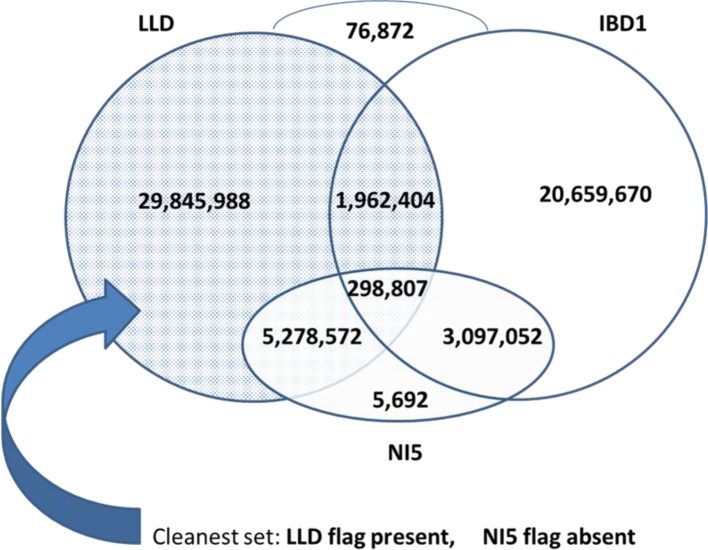
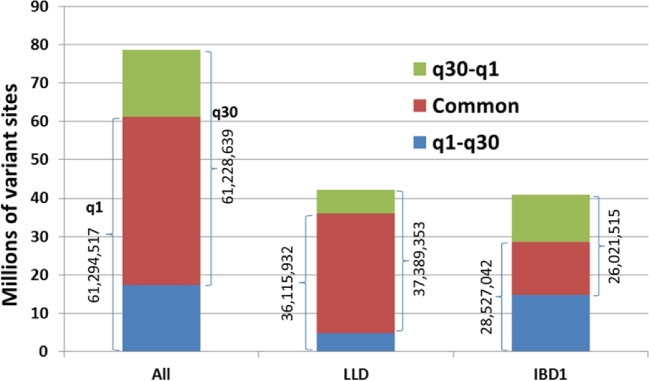
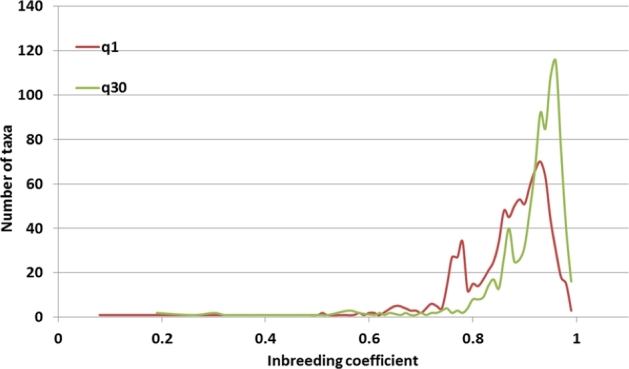
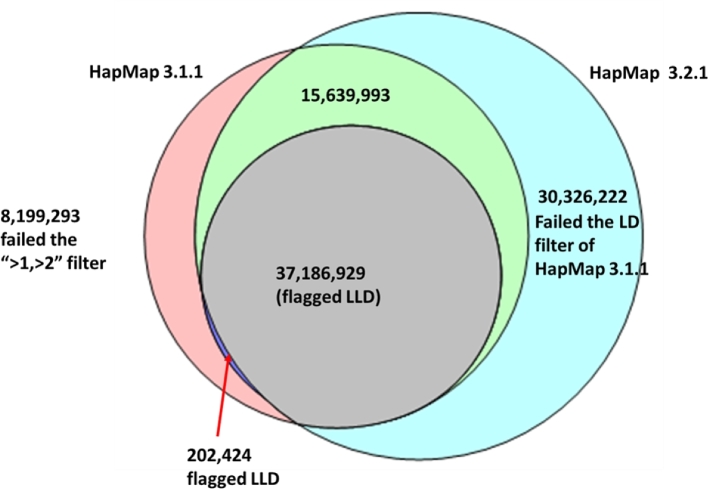
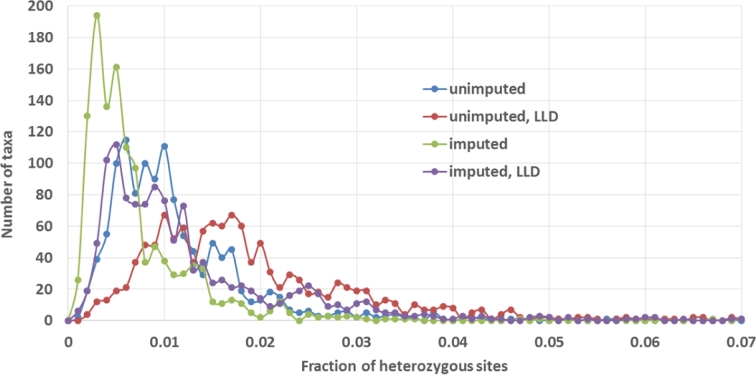
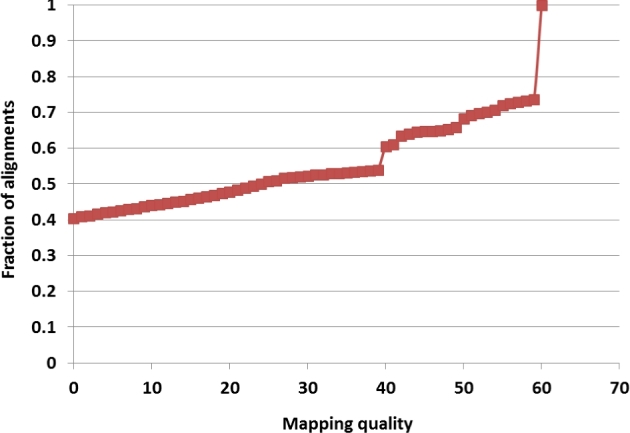
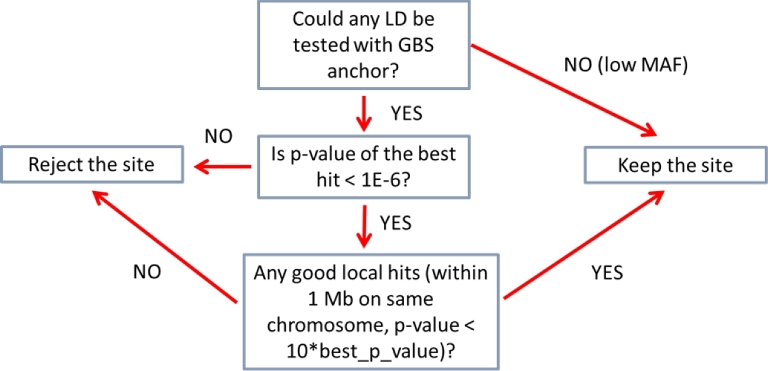
Results: A new computational pipeline was set up to process more than 12 trillion bp of sequencing data, and a set of population genetics filters was applied to identify more than 83 million variant sites.
Conclusions: We identified polymorphisms in regions where collinearity is largely preserved in the maize species. However, the fact that the B73 genome used as the reference only represents a fraction of all haplotypes is still an important limiting factor.
Figures
References
-
- Chia J-M, Song C, Bradbury PJ et al. . Maize HapMap2 identifies extant variation from a genome in flux. Nat Genet 2012;44:803–7. - PubMed
-
- Bukowski R, Guo X, Yanli Lu et al. . Supporting data for “Construction of the third generation Zea mays haplotype map.” GigaScience Database 2017. http://dx.doi.org/10.5524/100339. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
