

Biased signalling: from simple switches to allosteric microprocessors
- PMID: 29302067
- PMCID: PMC5936084
- DOI: 10.1038/nrd.2017.229
Biased signalling: from simple switches to allosteric microprocessors
Abstract
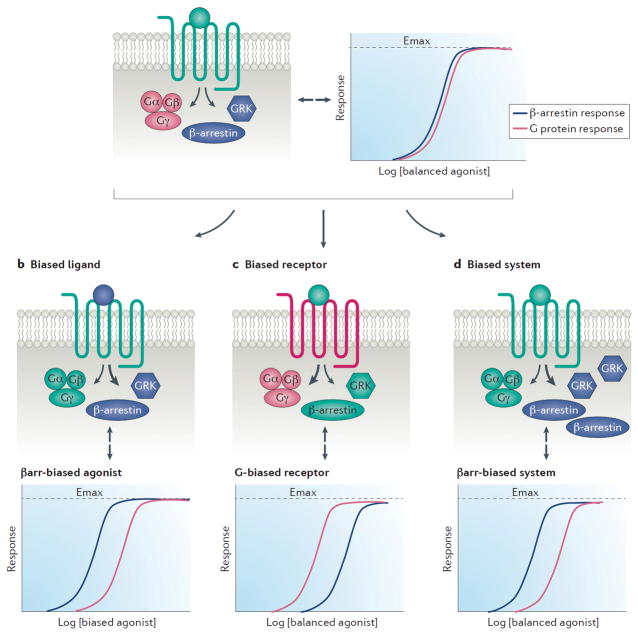
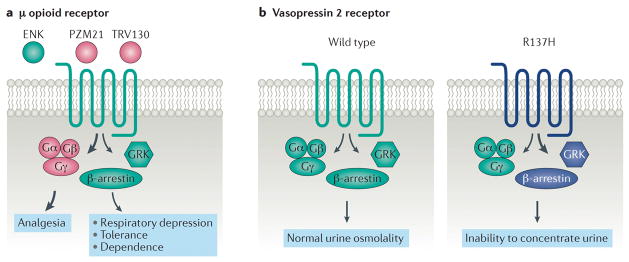
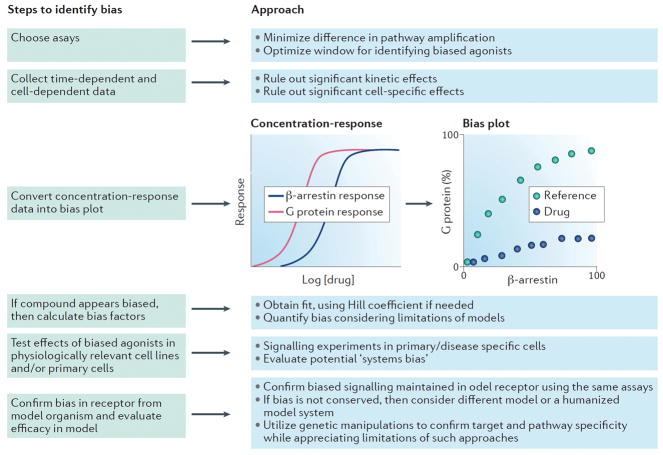
G protein-coupled receptors (GPCRs) are the largest class of receptors in the human genome and some of the most common drug targets. It is now well established that GPCRs can signal through multiple transducers, including heterotrimeric G proteins, GPCR kinases and β-arrestins. While these signalling pathways can be activated or blocked by 'balanced' agonists or antagonists, they can also be selectively activated in a 'biased' response. Biased responses can be induced by biased ligands, biased receptors or system bias, any of which can result in preferential signalling through G proteins or β-arrestins. At many GPCRs, signalling events mediated by G proteins and β-arrestins have been shown to have distinct biochemical and physiological actions from one another, and an accurate evaluation of biased signalling from pharmacology through physiology is crucial for preclinical drug development. Recent structural studies have provided snapshots of GPCR-transducer complexes, which should aid in the structure-based design of novel biased therapies. Our understanding of GPCRs has evolved from that of two-state, on-and-off switches to that of multistate allosteric microprocessors, in which biased ligands transmit distinct structural information that is processed into distinct biological outputs. The development of biased ligands as therapeutics heralds an era of increased drug efficacy with reduced drug side effects.
Conflict of interest statement
R.J.L. is a cofounder and shareholder of Trevena.
Figures
References
-
- Benovic JL, Strasser RH, Caron MG, Lefkowitz RJ. Beta-adrenergic receptor kinase: identification of a novel protein kinase that phosphorylates the agonist-occupied form of the receptor. Proceedings of the National Academy of Sciences of the United States of America. 1986;83:2797–2801. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
