

Unified understanding of folding and binding mechanisms of globular and intrinsically disordered proteins
- PMID: 29307002
- PMCID: PMC5899706
- DOI: 10.1007/s12551-017-0346-7
Unified understanding of folding and binding mechanisms of globular and intrinsically disordered proteins
Abstract
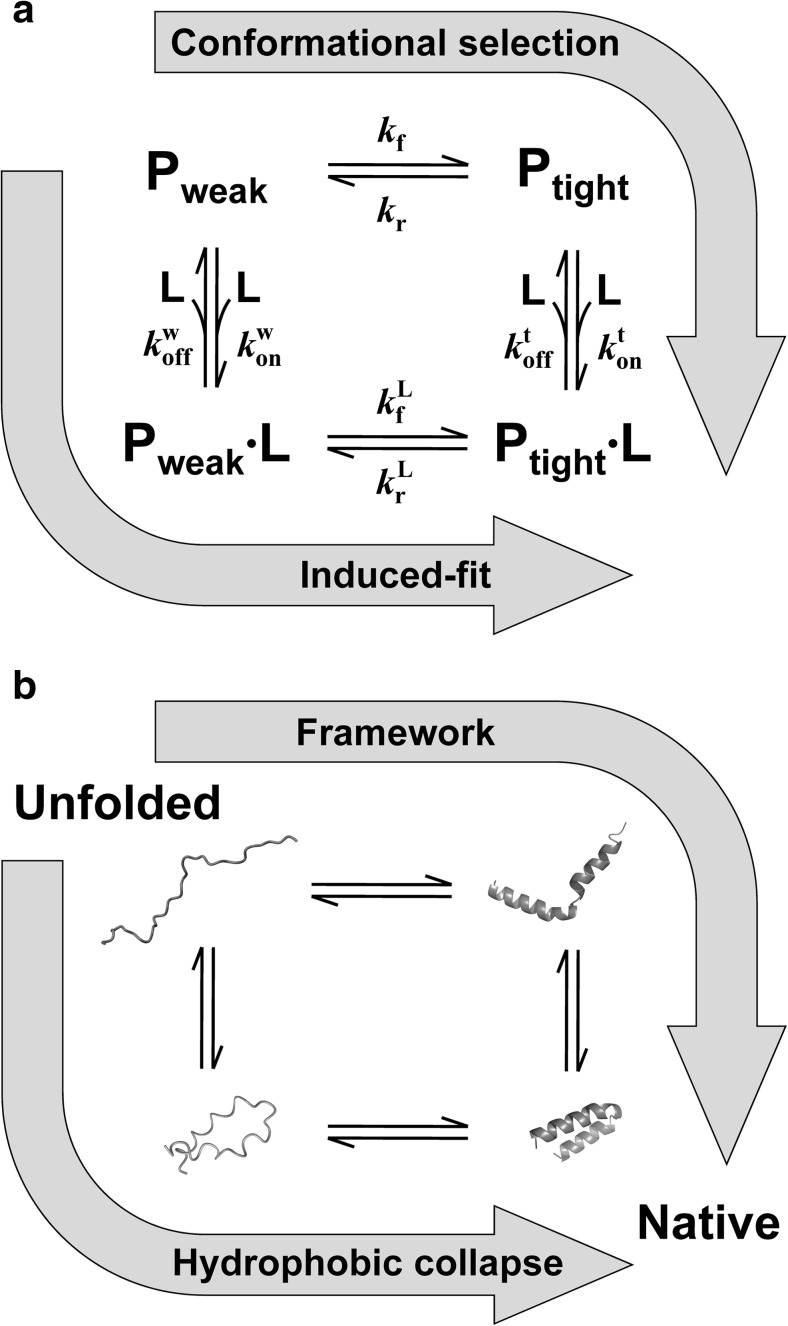
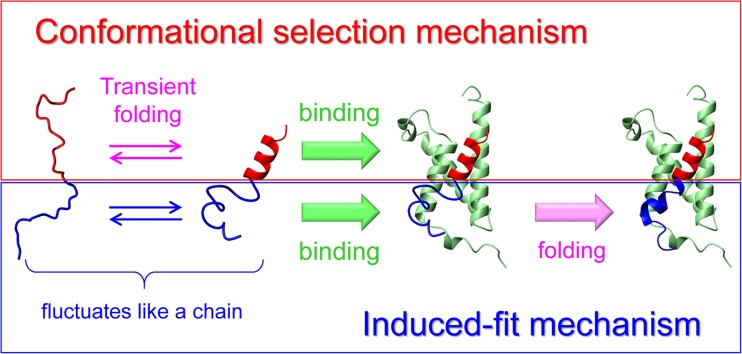
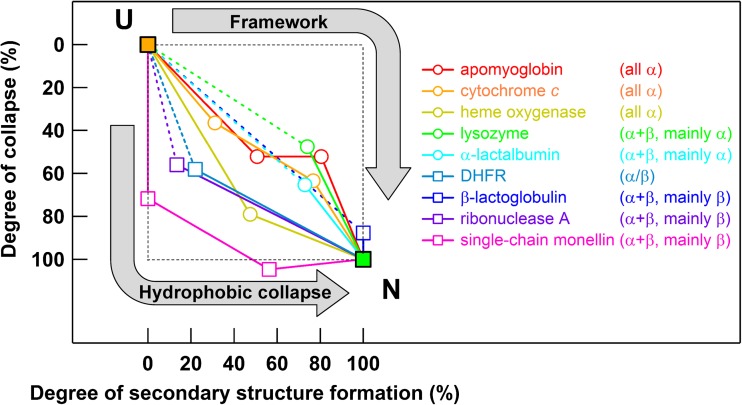
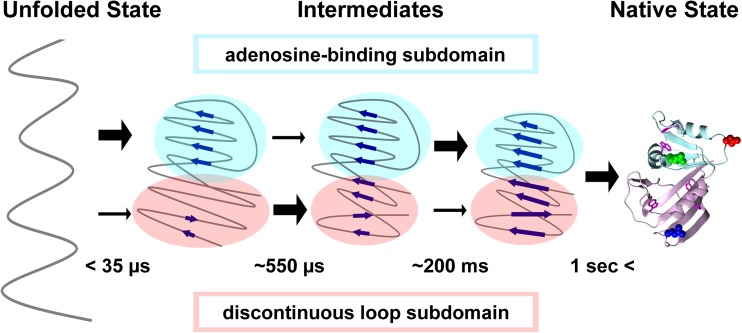
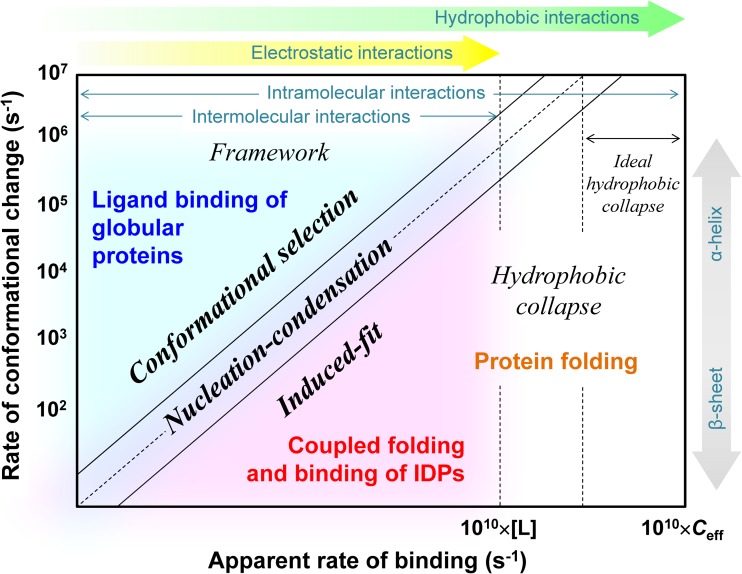
Extensive experimental and theoretical studies have advanced our understanding of the mechanisms of folding and binding of globular proteins, and coupled folding and binding of intrinsically disordered proteins (IDPs). The forces responsible for conformational changes and binding are common in both proteins; however, these mechanisms have been separately discussed. Here, we attempt to integrate the mechanisms of coupled folding and binding of IDPs, folding of small and multi-subdomain proteins, folding of multimeric proteins, and ligand binding of globular proteins in terms of conformational selection and induced-fit mechanisms as well as the nucleation-condensation mechanism that is intermediate between them. Accumulating evidence has shown that both the rate of conformational change and apparent rate of binding between interacting elements can determine reaction mechanisms. Coupled folding and binding of IDPs occurs mainly by induced-fit because of the slow folding in the free form, while ligand binding of globular proteins occurs mainly by conformational selection because of rapid conformational change. Protein folding can be regarded as the binding of intramolecular segments accompanied by secondary structure formation. Multi-subdomain proteins fold mainly by the induced-fit (hydrophobic collapse) mechanism, as the connection of interacting segments enhances the binding (compaction) rate. Fewer hydrophobic residues in small proteins reduce the intramolecular binding rate, resulting in the nucleation-condensation mechanism. Thus, the folding and binding of globular proteins and IDPs obey the same general principle, suggesting that the coarse-grained, statistical mechanical model of protein folding is promising for a unified theoretical description of all mechanisms.
Keywords: Conformational selection; Induced-fit; Intrinsically disordered protein; Ligand binding; Nucleation–condensation; Protein folding.
Conflict of interest statement
Funding
This study was funded by Grants-in-Aids for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (MEXT).
Conflict of interest
Munehito Arai declares that he has no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by the author.
Figures
References
-
- Akiyama S, Takahashi S, Ishimori K, Morishima I. Stepwise formation of α-helices during cytochrome c folding. Nat Struct Biol. 2000;7:514–520. - PubMed
-
- Arai M, Hamel P, Kanaya E, Inaka K, Miki K, Kikuchi M, Kuwajima K. Effect of an alternative disulfide bond on the structure, stability, and folding of human lysozyme. Biochemistry. 2000;39:3472–3479. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
