

Distinct temporal roles for the promyelocytic leukaemia (PML) protein in the sequential regulation of intracellular host immunity to HSV-1 infection
- PMID: 29309427
- PMCID: PMC5757968
- DOI: 10.1371/journal.ppat.1006769
Distinct temporal roles for the promyelocytic leukaemia (PML) protein in the sequential regulation of intracellular host immunity to HSV-1 infection
Erratum in
-
Correction: Distinct temporal roles for the promyelocytic leukaemia (PML) protein in the sequential regulation of intracellular host immunity to HSV-1 infection.PLoS Pathog. 2018 Feb 22;14(2):e1006927. doi: 10.1371/journal.ppat.1006927. eCollection 2018 Feb. PLoS Pathog. 2018. PMID: 29470515 Free PMC article.
Abstract
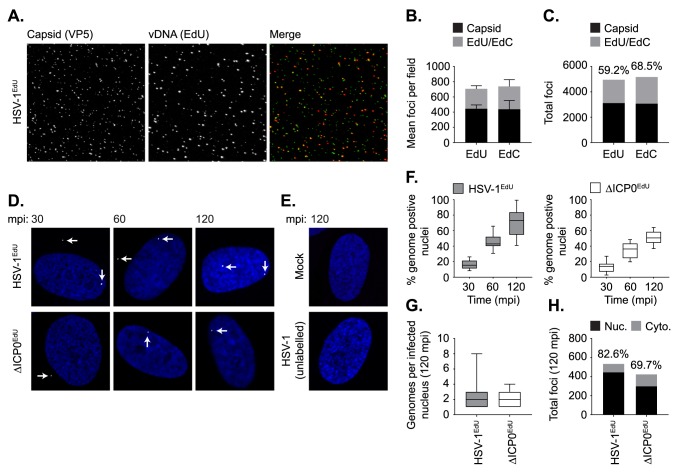
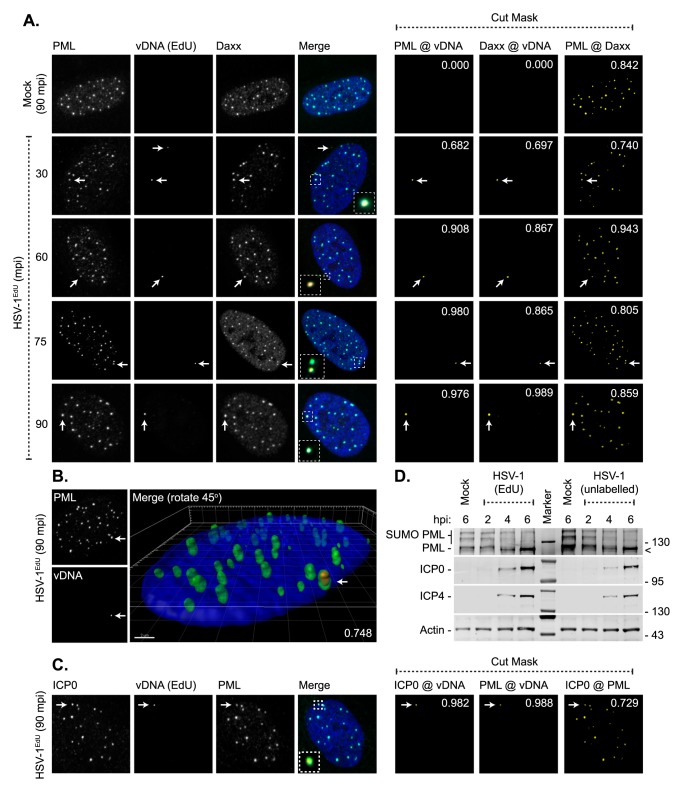
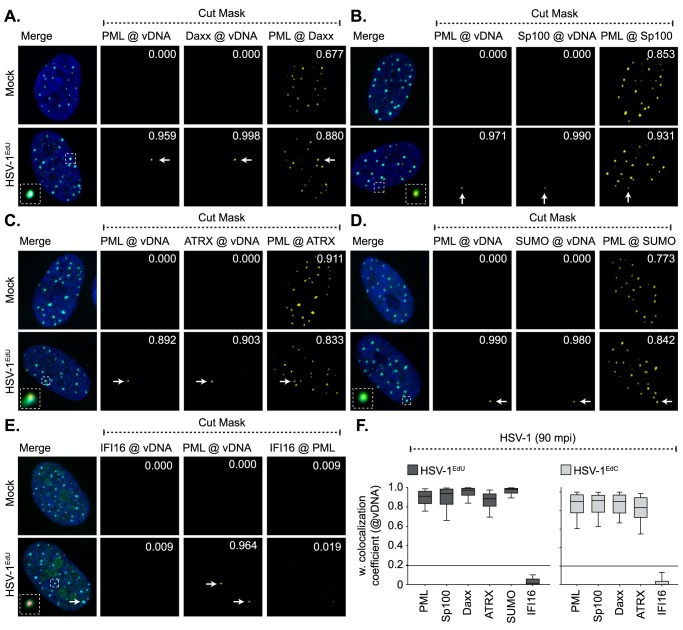
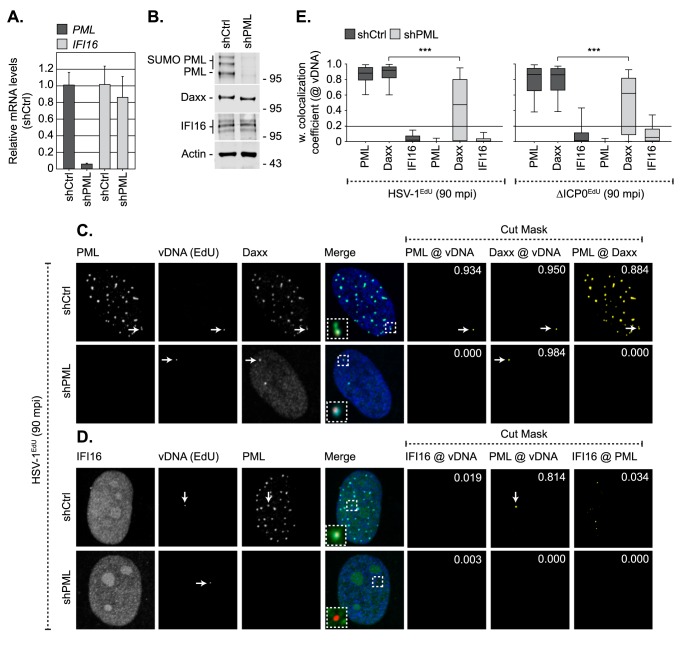
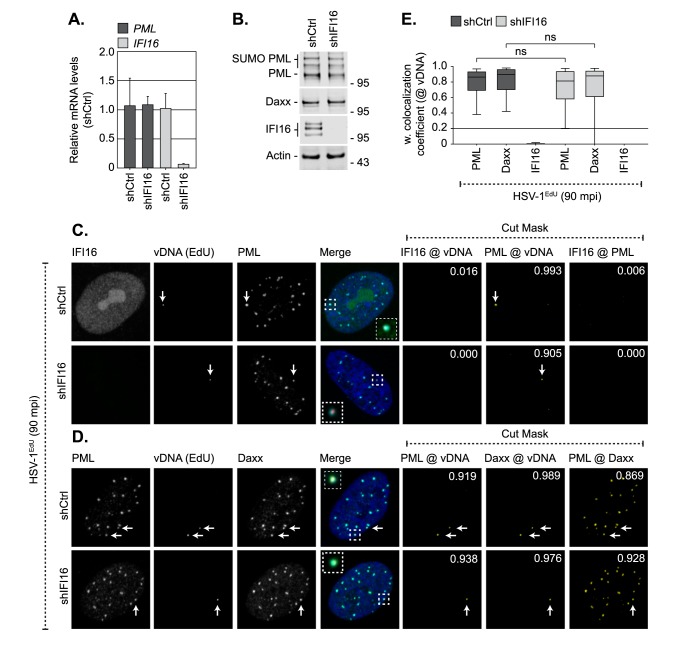
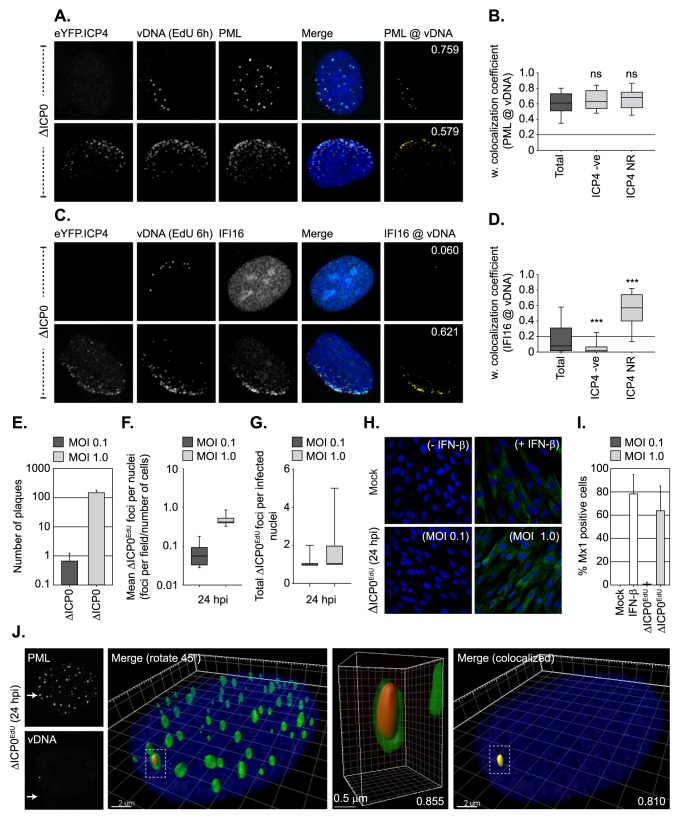
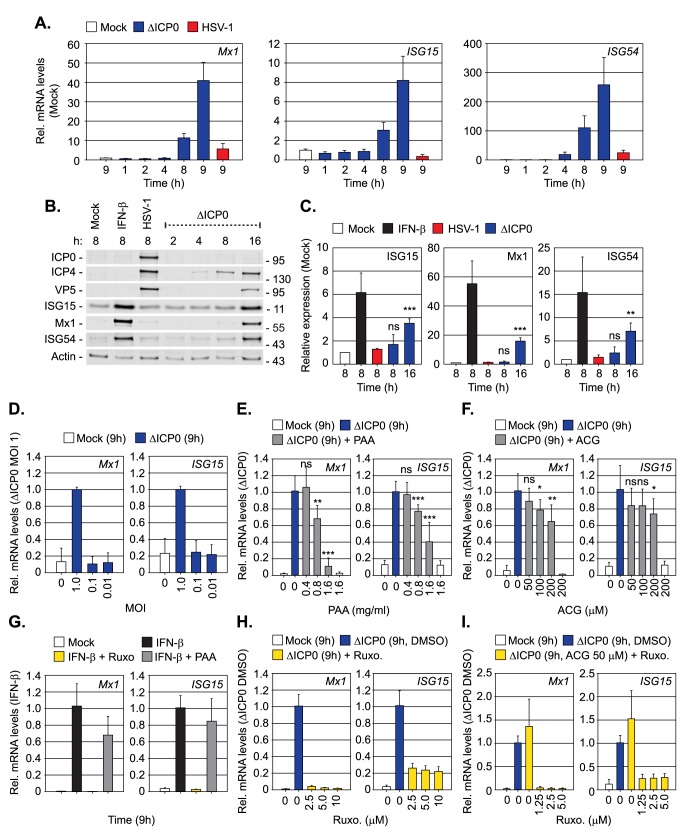
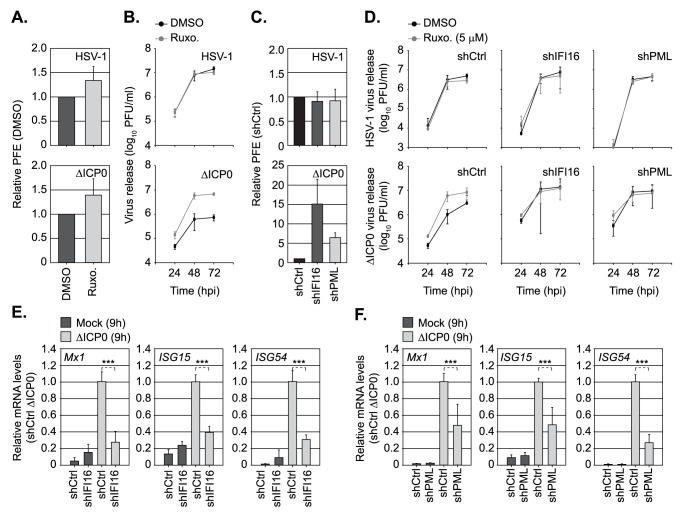
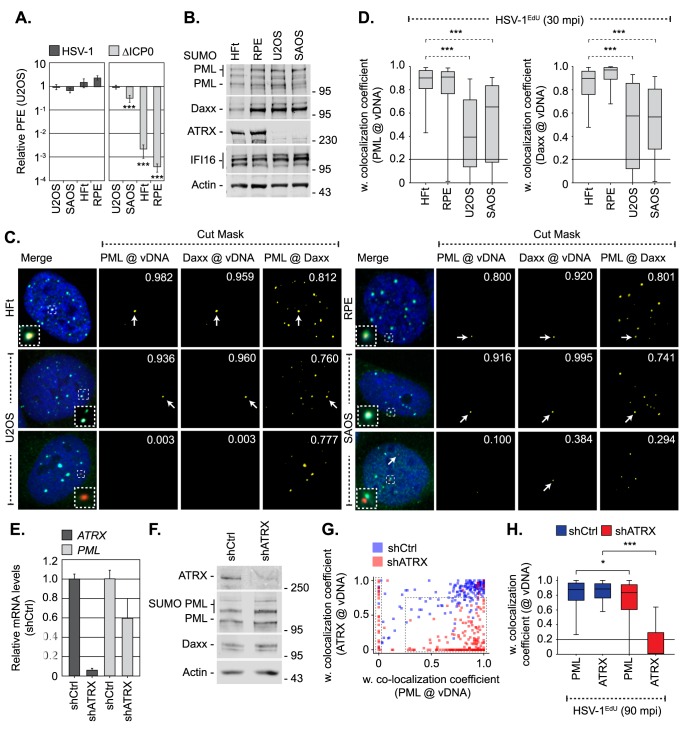
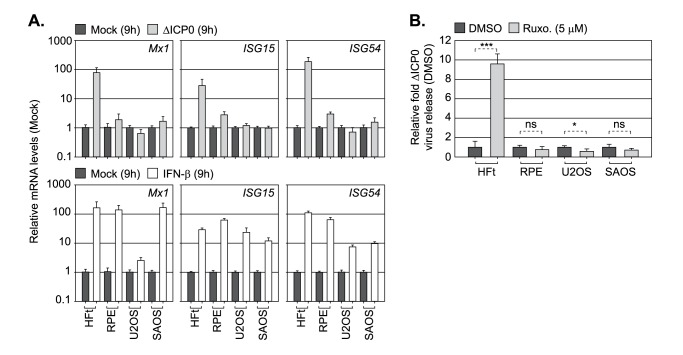
Detection of viral nucleic acids plays a critical role in the induction of intracellular host immune defences. However, the temporal recruitment of immune regulators to infecting viral genomes remains poorly defined due to the technical difficulties associated with low genome copy-number detection. Here we utilize 5-Ethynyl-2'-deoxyuridine (EdU) labelling of herpes simplex virus 1 (HSV-1) DNA in combination with click chemistry to examine the sequential recruitment of host immune regulators to infecting viral genomes under low multiplicity of infection conditions. Following viral genome entry into the nucleus, PML-nuclear bodies (PML-NBs) rapidly entrapped viral DNA (vDNA) leading to a block in viral replication in the absence of the viral PML-NB antagonist ICP0. This pre-existing intrinsic host defence to infection occurred independently of the vDNA pathogen sensor IFI16 (Interferon Gamma Inducible Protein 16) and the induction of interferon stimulated gene (ISG) expression, demonstrating that vDNA entry into the nucleus alone is not sufficient to induce a robust innate immune response. Saturation of this pre-existing intrinsic host defence during HSV-1 ICP0-null mutant infection led to the stable recruitment of PML and IFI16 into vDNA complexes associated with ICP4, and led to the induction of ISG expression. This induced innate immune response occurred in a PML-, IFI16-, and Janus-Associated Kinase (JAK)-dependent manner and was restricted by phosphonoacetic acid, demonstrating that vDNA polymerase activity is required for the robust induction of ISG expression during HSV-1 infection. Our data identifies dual roles for PML in the sequential regulation of intrinsic and innate immunity to HSV-1 infection that are dependent on viral genome delivery to the nucleus and the onset of vDNA replication, respectively. These intracellular host defences are counteracted by ICP0, which targets PML for degradation from the outset of nuclear infection to promote vDNA release from PML-NBs and the onset of HSV-1 lytic replication.
Conflict of interest statement
The authors declare no competing interests
Figures
References
-
- Komatsu T, Nagata K, Wodrich H. The Role of Nuclear Antiviral Factors against Invading DNA Viruses: The Immediate Fate of Incoming Viral Genomes. Viruses. 2016;8(10). doi: 10.3390/v8100290 ; PubMed Central PMCID: PMCPMC5086622. - DOI - PMC - PubMed
-
- Yan N, Chen ZJ. Intrinsic antiviral immunity. Nat Immunol. 2012;13(3):214–22. doi: 10.1038/ni.2229 ; PubMed Central PMCID: PMCPMC3549670. - DOI - PMC - PubMed
-
- Blanco-Melo D, Venkatesh S, Bieniasz PD. Intrinsic cellular defenses against human immunodeficiency viruses. Immunity. 2012;37(3):399–411. Epub 2012/09/25. doi: 10.1016/j.immuni.2012.08.013 [pii]. . - DOI - PMC - PubMed
-
- Diner BA, Lum KK, Cristea IM. The emerging role of nuclear viral DNA sensors. J Biol Chem. 2015;290(44):26412–21. doi: 10.1074/jbc.R115.652289 ; PubMed Central PMCID: PMCPMC4646299. - DOI - PMC - PubMed
-
- Knipe DM. Nuclear sensing of viral DNA, epigenetic regulation of herpes simplex virus infection, and innate immunity. Virology. 2015;479–480:153–9. doi: 10.1016/j.virol.2015.02.009 ; PubMed Central PMCID: PMCPMC4424148. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
