

Epigenetic supersimilarity of monozygotic twin pairs
- PMID: 29310692
- PMCID: PMC5759268
- DOI: 10.1186/s13059-017-1374-0
Epigenetic supersimilarity of monozygotic twin pairs
Abstract
Background: Monozygotic twins have long been studied to estimate heritability and explore epigenetic influences on phenotypic variation. The phenotypic and epigenetic similarities of monozygotic twins have been assumed to be largely due to their genetic identity.
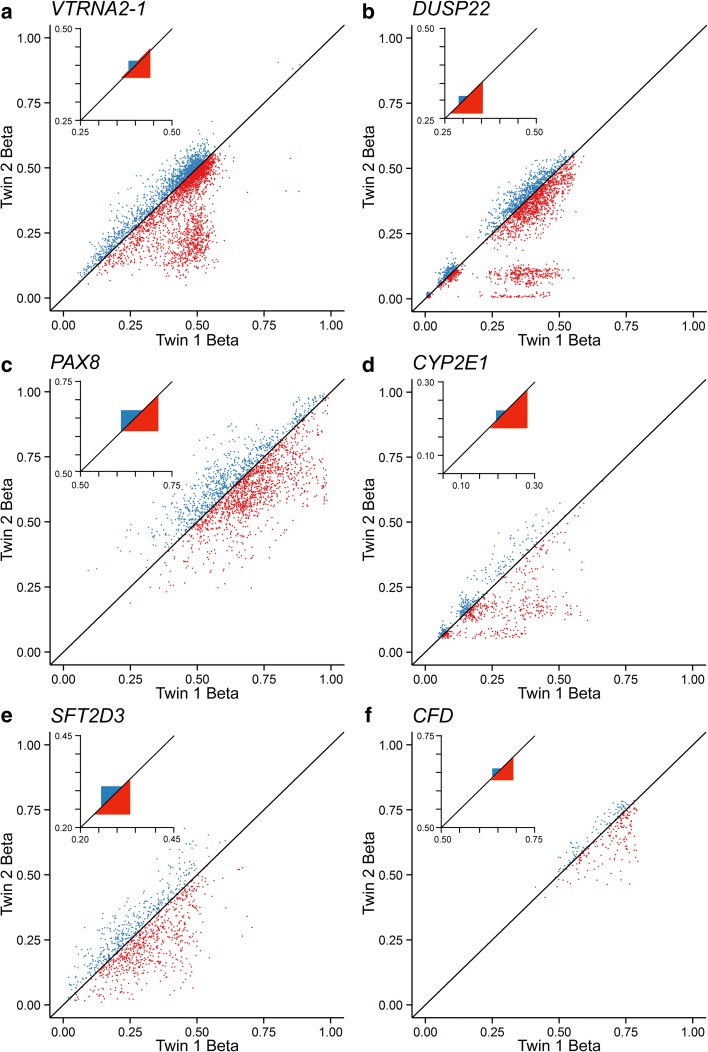
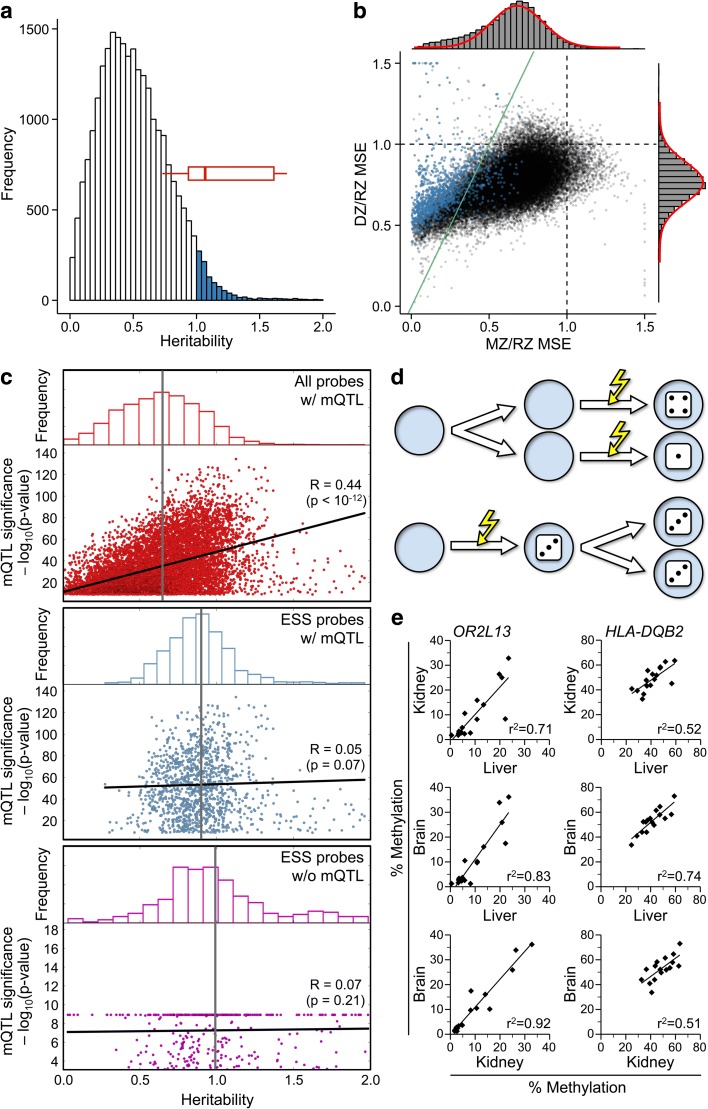
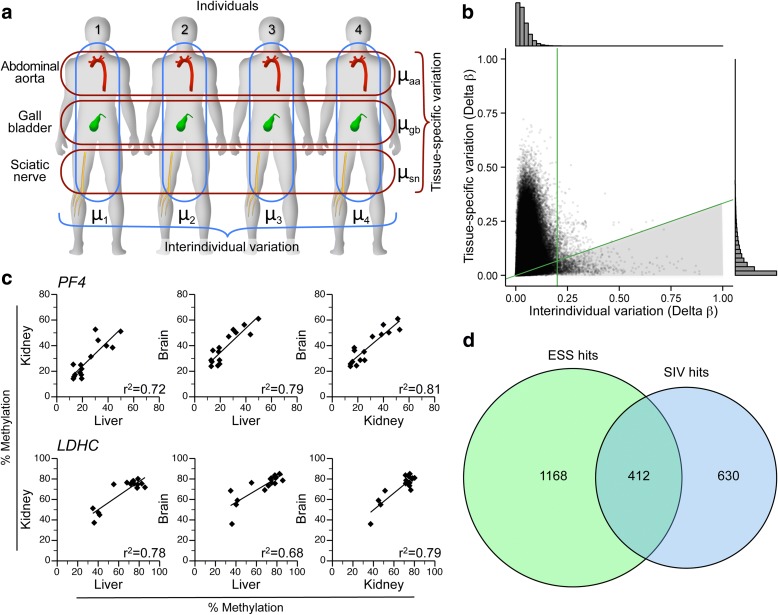
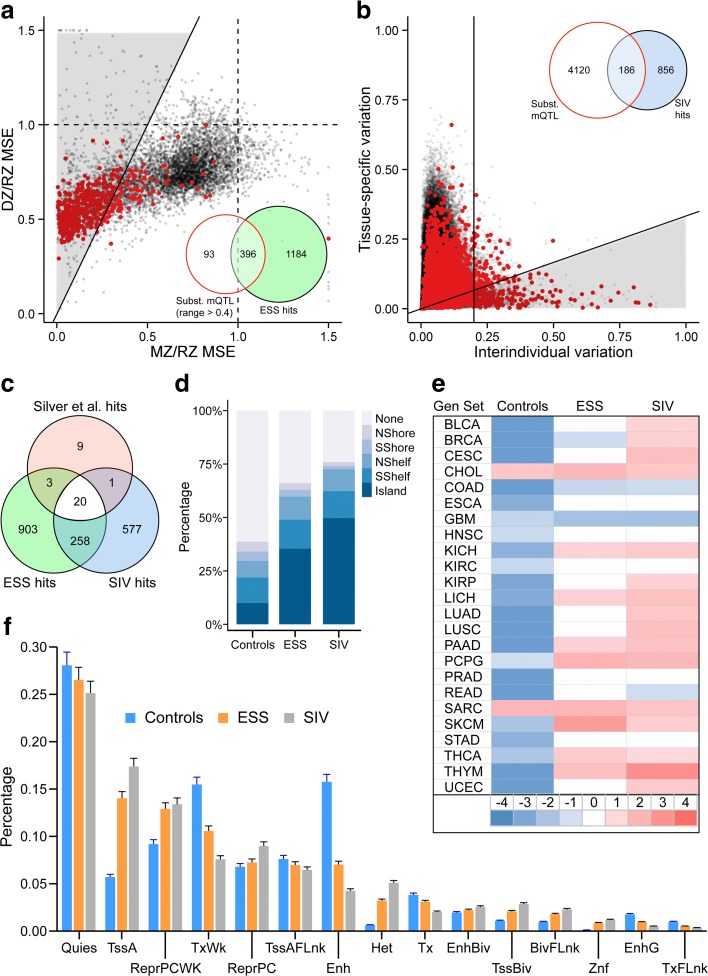
Results: Here, by analyzing data from a genome-scale study of DNA methylation in monozygotic and dizygotic twins, we identified genomic regions at which the epigenetic similarity of monozygotic twins is substantially greater than can be explained by their genetic identity. This "epigenetic supersimilarity" apparently results from locus-specific establishment of epigenotype prior to embryo cleavage during twinning. Epigenetically supersimilar loci exhibit systemic interindividual epigenetic variation and plasticity to periconceptional environment and are enriched in sub-telomeric regions. In case-control studies nested in a prospective cohort, blood DNA methylation at these loci years before diagnosis is associated with risk of developing several types of cancer.
Conclusions: These results establish a link between early embryonic epigenetic development and adult disease. More broadly, epigenetic supersimilarity is a previously unrecognized phenomenon that may contribute to the phenotypic similarity of monozygotic twins.
Keywords: Cancer; DOHaD; Developmental programming; Dizygotic; Epigenetics; Metastable epialleles; Monozygotic; Twins.
Conflict of interest statement
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
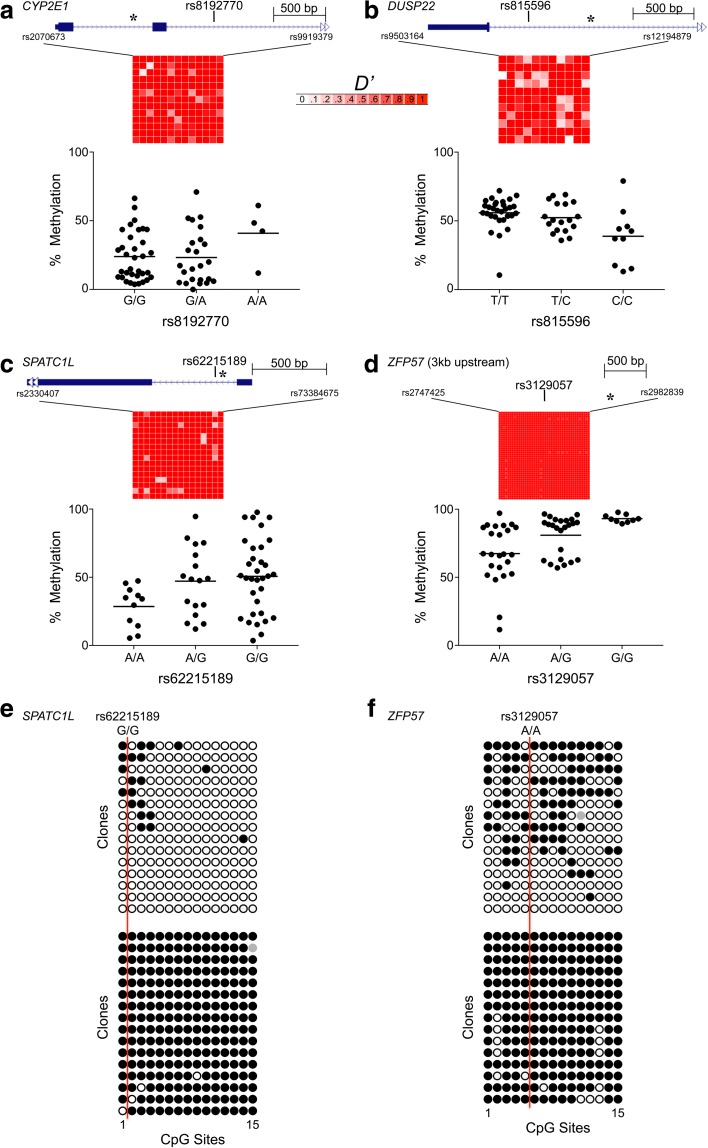
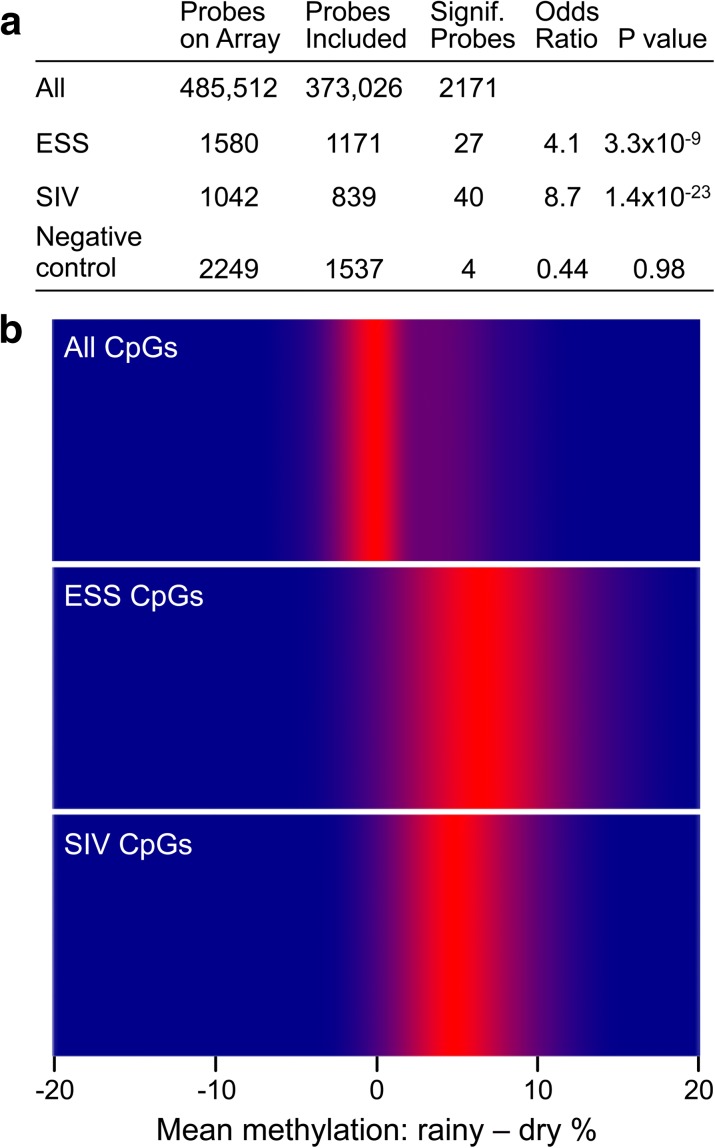
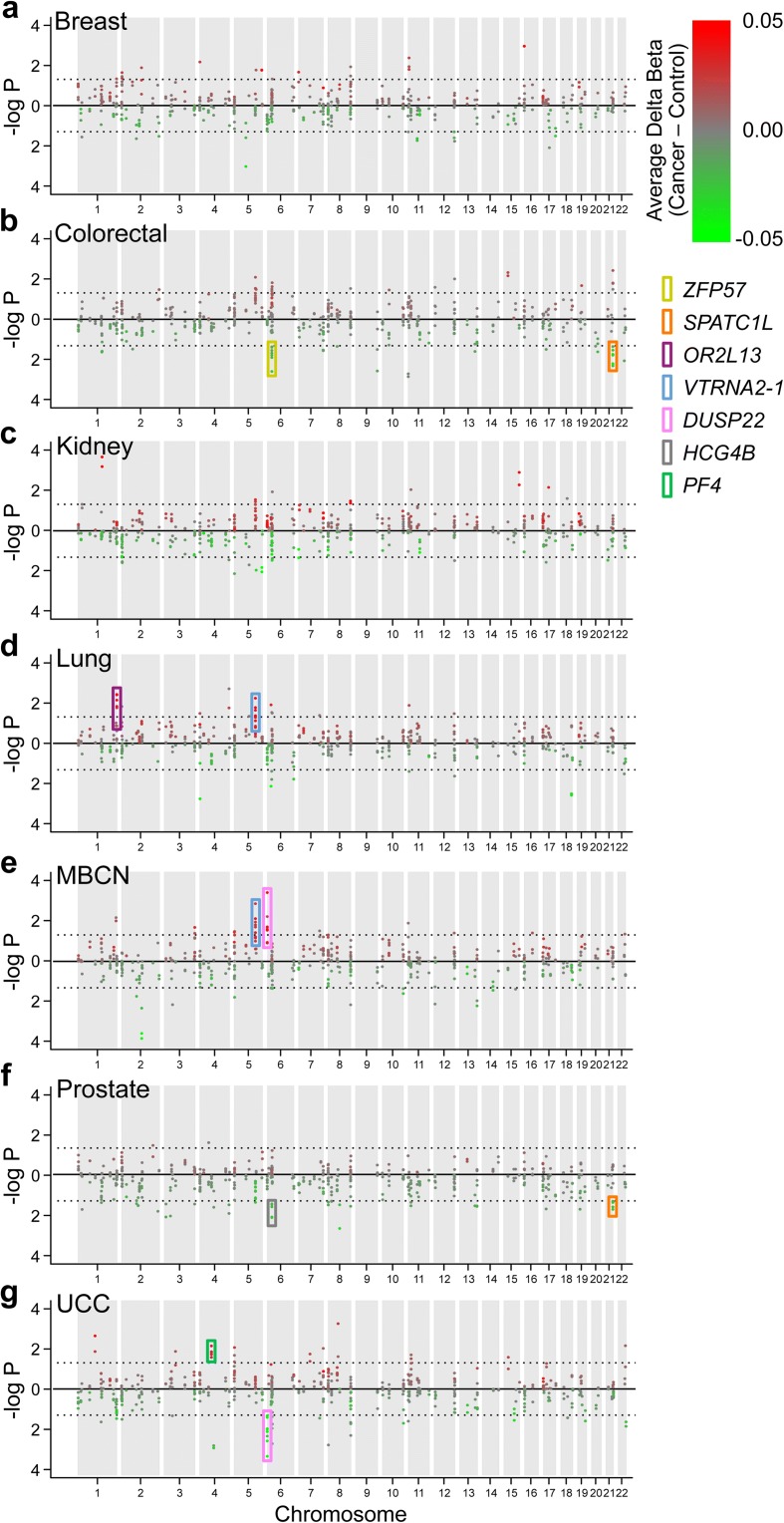
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
