

Dysregulation of cotranscriptional alternative splicing underlies CHARGE syndrome
- PMID: 29311329
- PMCID: PMC5789929
- DOI: 10.1073/pnas.1715378115
Dysregulation of cotranscriptional alternative splicing underlies CHARGE syndrome
Abstract
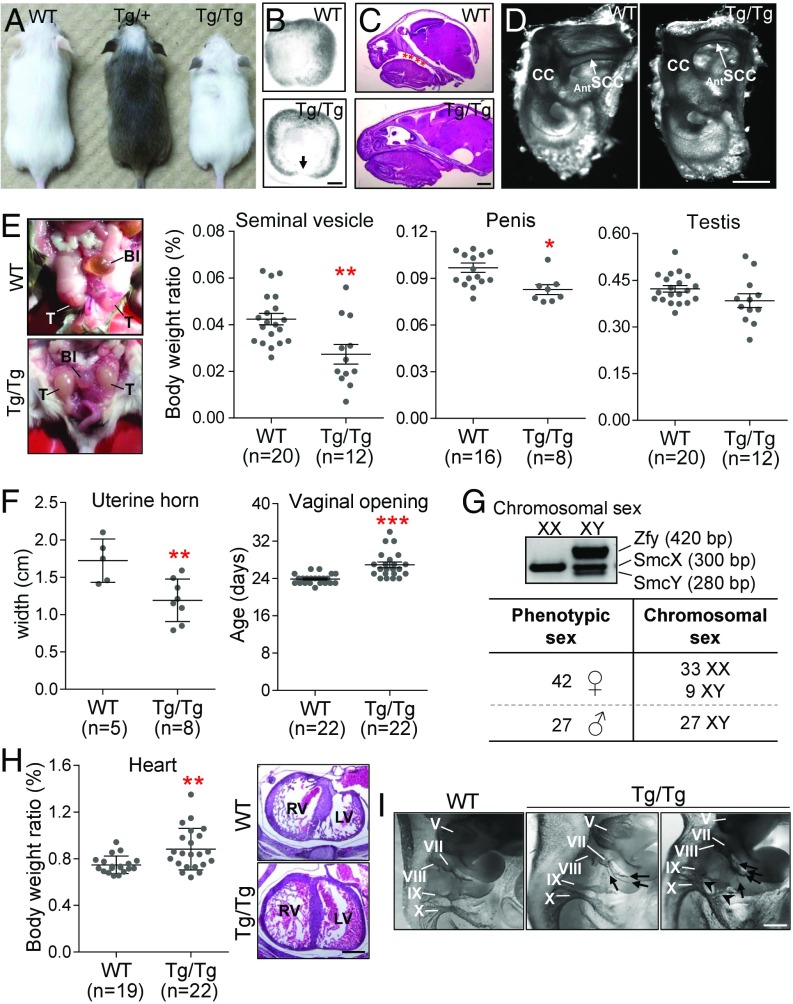
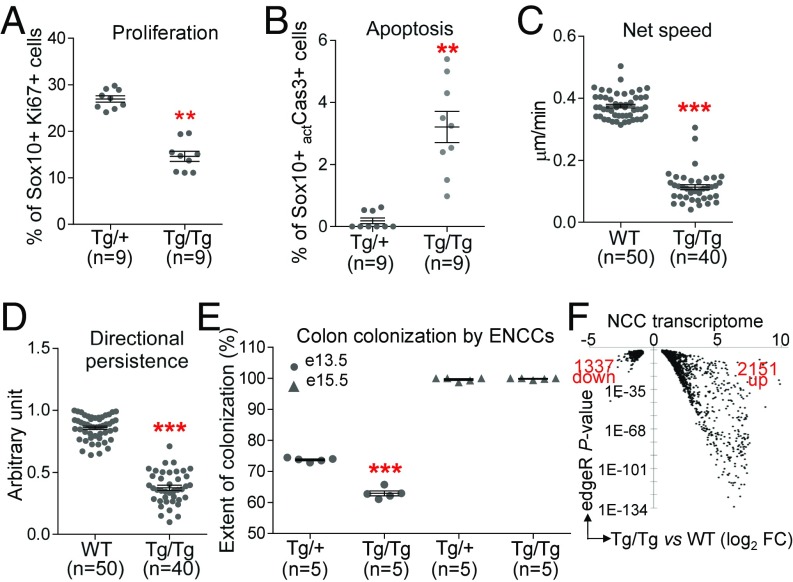
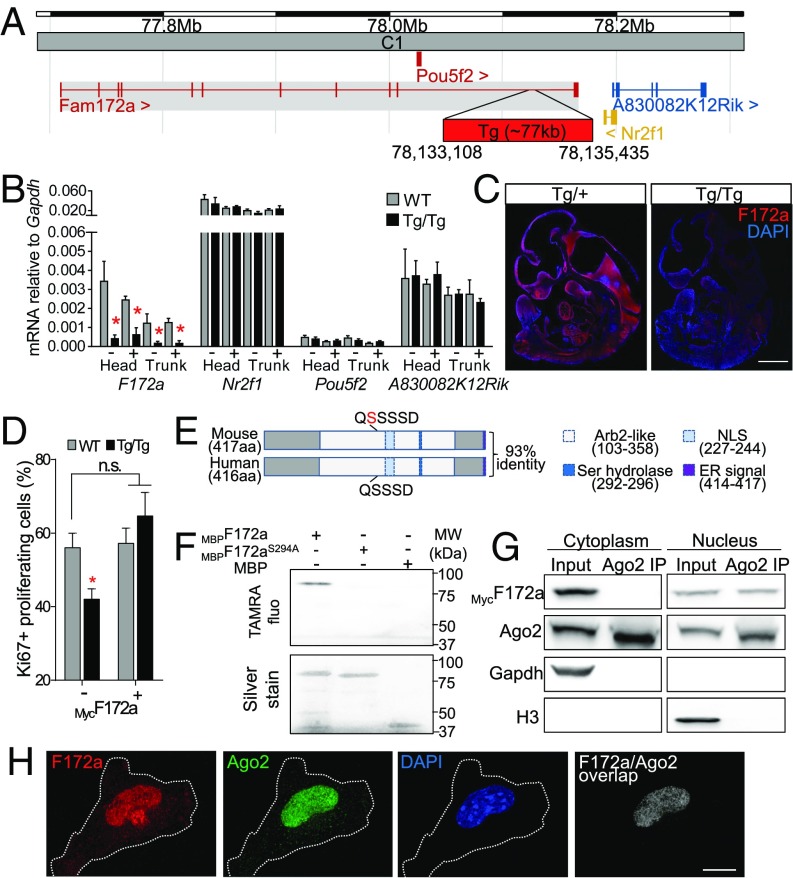
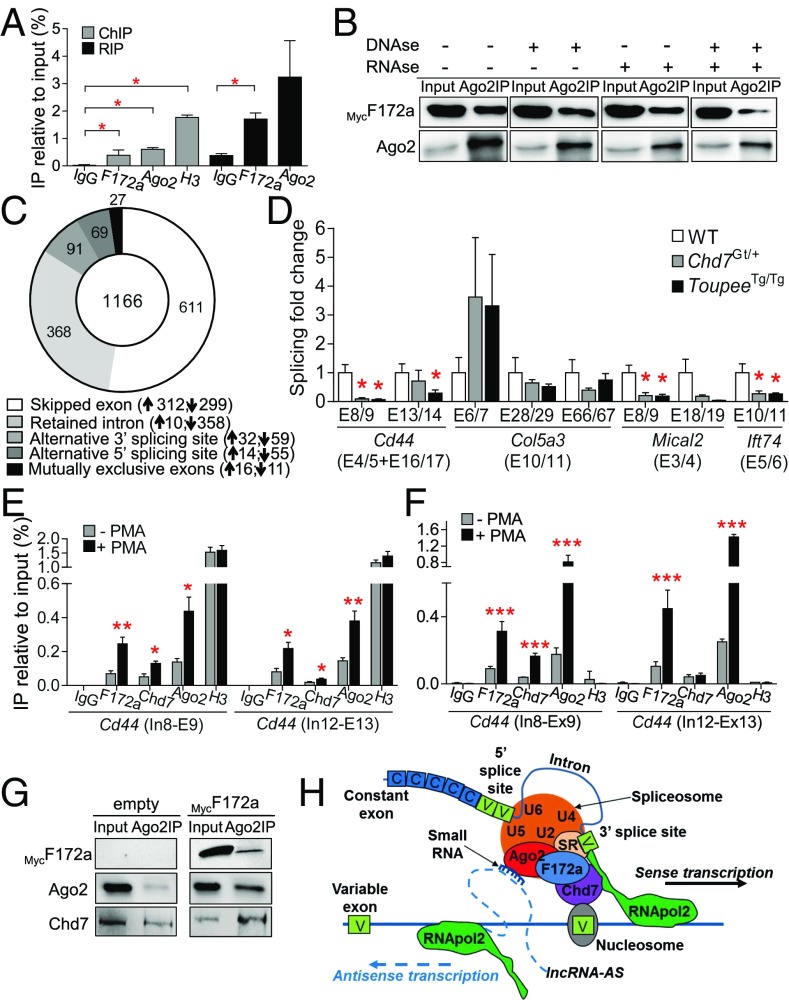
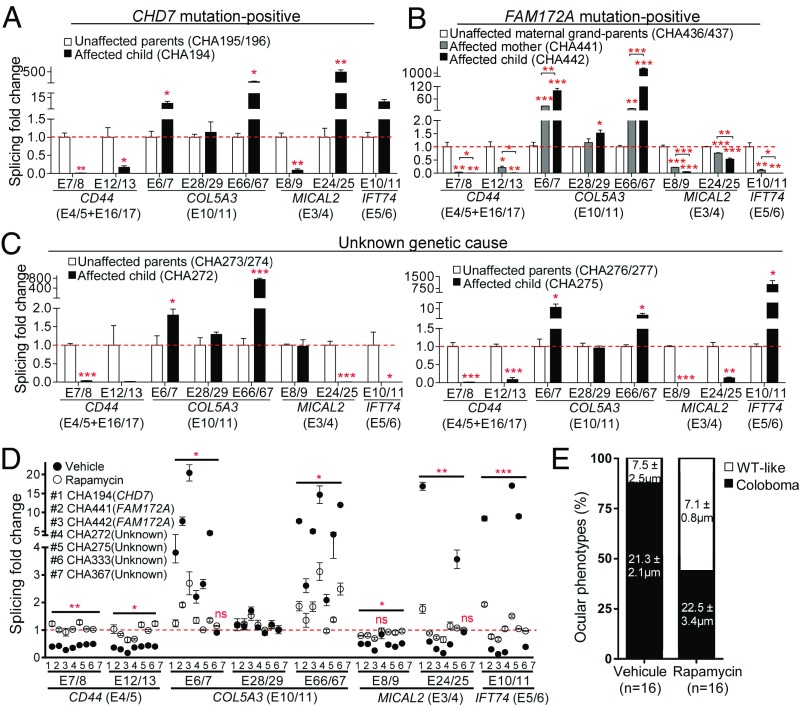
CHARGE syndrome-which stands for coloboma of the eye, heart defects, atresia of choanae, retardation of growth/development, genital abnormalities, and ear anomalies-is a severe developmental disorder with wide phenotypic variability, caused mainly by mutations in CHD7 (chromodomain helicase DNA-binding protein 7), known to encode a chromatin remodeler. The genetic lesions responsible for CHD7 mutation-negative cases are unknown, at least in part because the pathogenic mechanisms underlying CHARGE syndrome remain poorly defined. Here, we report the characterization of a mouse model for CHD7 mutation-negative cases of CHARGE syndrome generated by insertional mutagenesis of Fam172a (family with sequence similarity 172, member A). We show that Fam172a plays a key role in the regulation of cotranscriptional alternative splicing, notably by interacting with Ago2 (Argonaute-2) and Chd7. Validation studies in a human cohort allow us to propose that dysregulation of cotranscriptional alternative splicing is a unifying pathogenic mechanism for both CHD7 mutation-positive and CHD7 mutation-negative cases. We also present evidence that such splicing defects can be corrected in vitro by acute rapamycin treatment.
Keywords: CHARGE syndrome; Fam172a; alternative splicing; neural crest cells; sex reversal.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Hsu P, et al. CHARGE syndrome: A review. J Paediatr Child Health. 2014;50:504–511. - PubMed
-
- Hale CL, Niederriter AN, Green GE, Martin DM. Response to correspondence to Hale et al. atypical phenotypes associated with pathogenic CHD7 variants and a proposal for broadening CHARGE syndrome clinical diagnostic criteria. Am J Med Genet A. 2016;170:3367–3368. - PubMed
-
- Delahaye A, et al. Familial CHARGE syndrome because of CHD7 mutation: Clinical intra- and interfamilial variability. Clin Genet. 2007;72:112–121. - PubMed
-
- Jongmans MC, et al. Familial CHARGE syndrome and the CHD7 gene: A recurrent missense mutation, intrafamilial recurrence and variability. Am J Med Genet A. 2008;146A:43–50. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
