

Translating genomics into practice for real-time surveillance and response to carbapenemase-producing Enterobacteriaceae: evidence from a complex multi-institutional KPC outbreak
- PMID: 29312831
- PMCID: PMC5756455
- DOI: 10.7717/peerj.4210
Translating genomics into practice for real-time surveillance and response to carbapenemase-producing Enterobacteriaceae: evidence from a complex multi-institutional KPC outbreak
Abstract
Background: Until recently, Klebsiella pneumoniae carbapenemase (KPC)-producing Enterobacteriaceae were rarely identified in Australia. Following an increase in the number of incident cases across the state of Victoria, we undertook a real-time combined genomic and epidemiological investigation. The scope of this study included identifying risk factors and routes of transmission, and investigating the utility of genomics to enhance traditional field epidemiology for informing management of established widespread outbreaks.
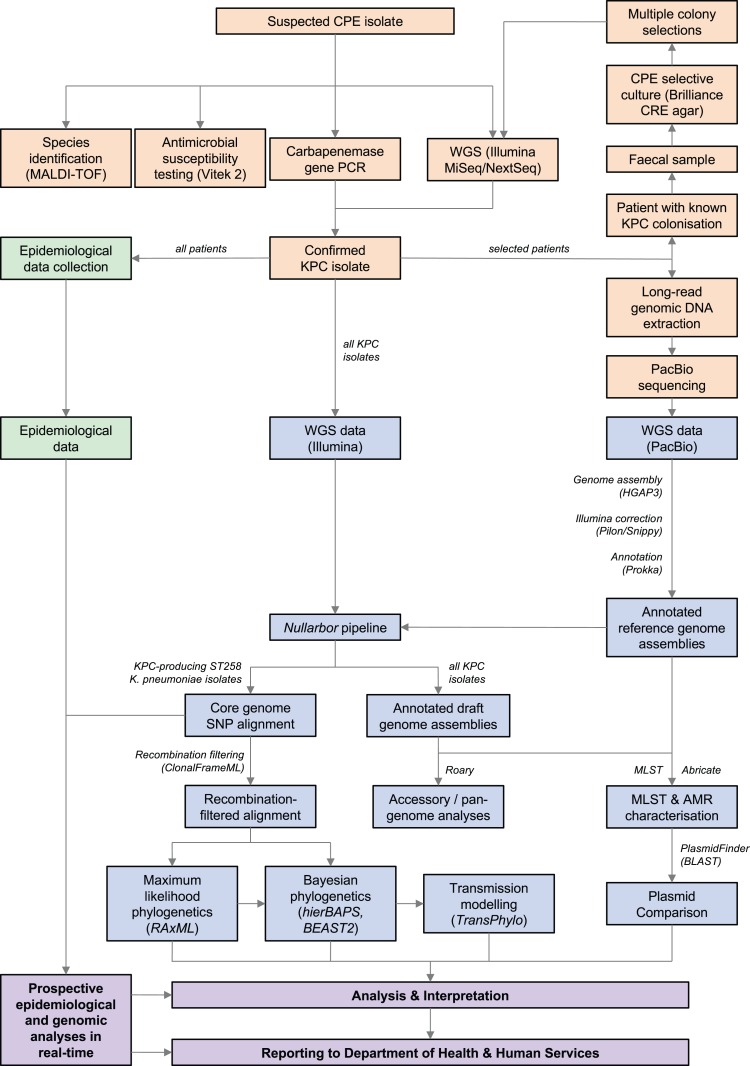
Methods: All KPC-producing Enterobacteriaceae isolates referred to the state reference laboratory from 2012 onwards were included. Whole-genome sequencing was performed in parallel with a detailed descriptive epidemiological investigation of each case, using Illumina sequencing on each isolate. This was complemented with PacBio long-read sequencing on selected isolates to establish high-quality reference sequences and interrogate characteristics of KPC-encoding plasmids.
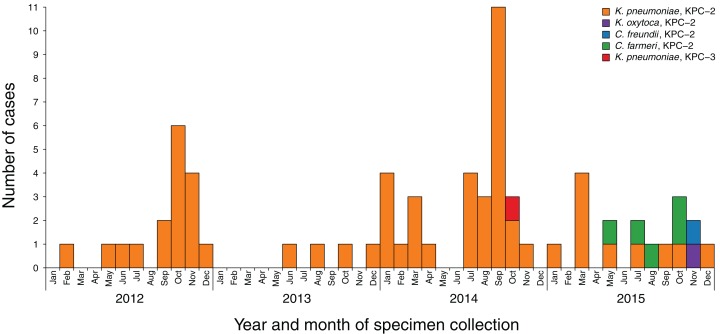
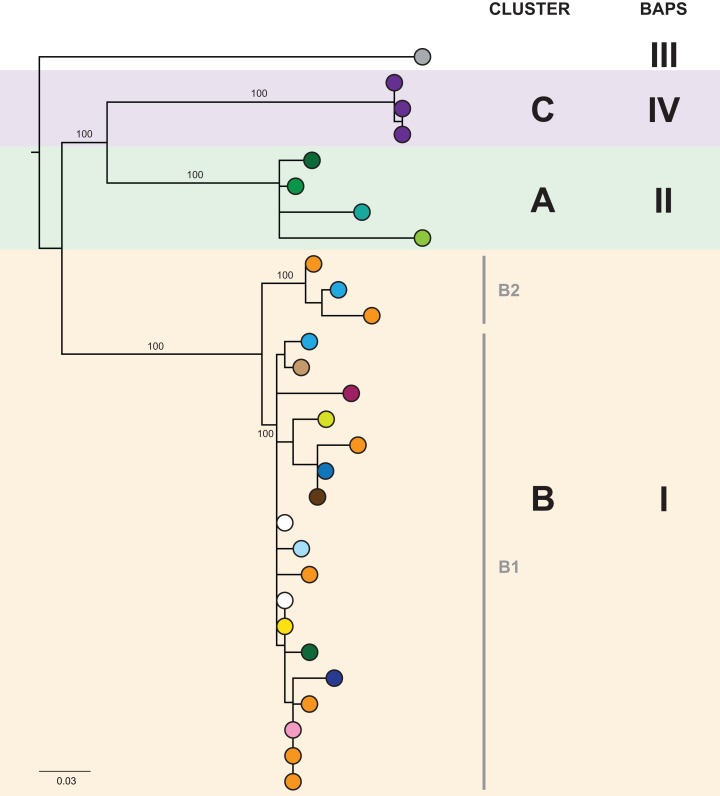
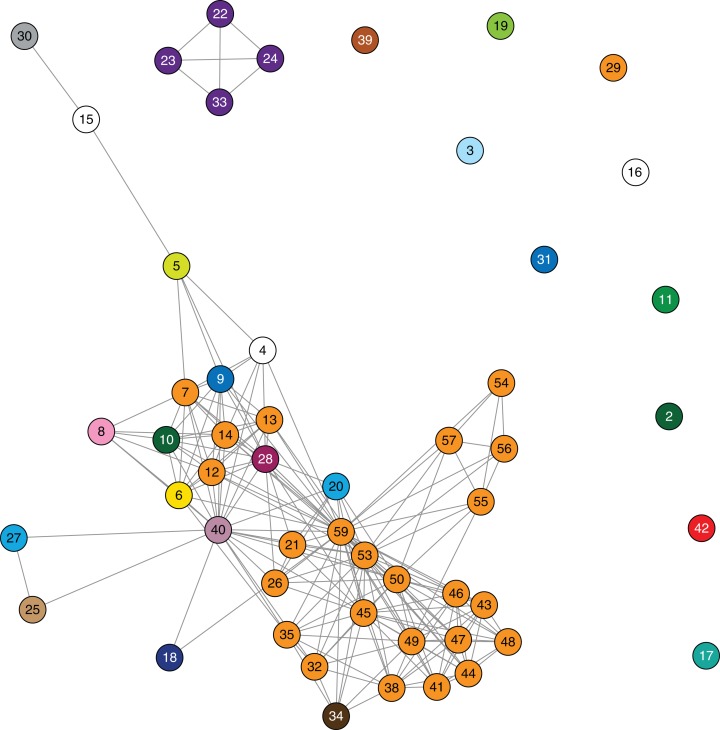
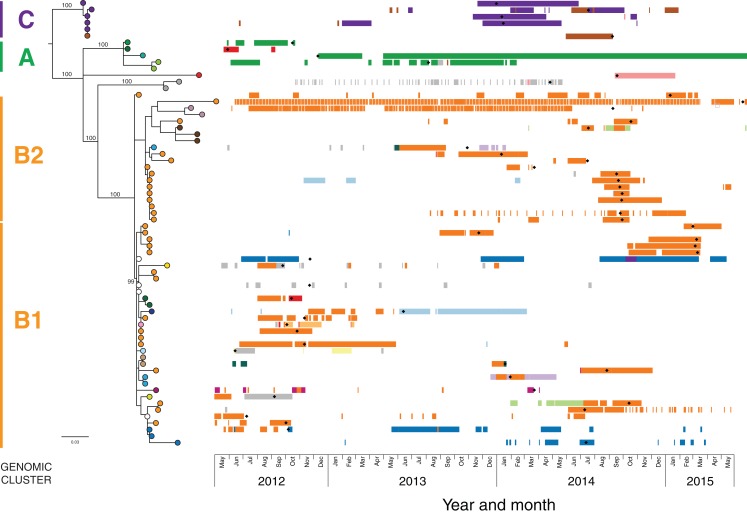
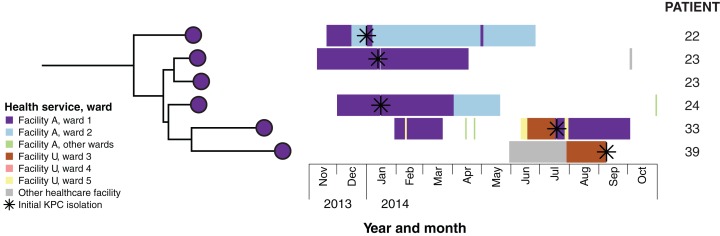
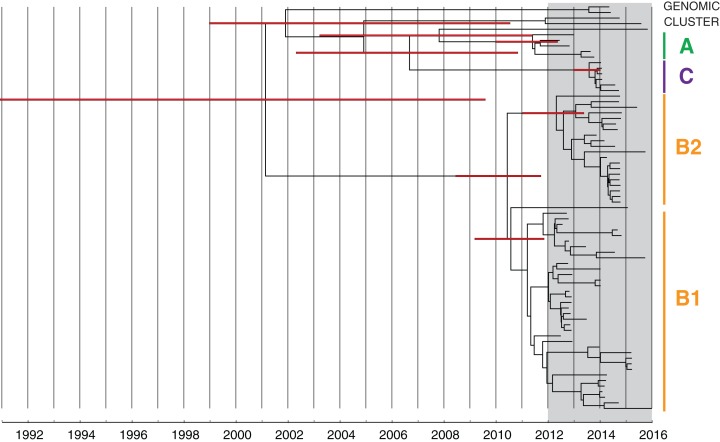
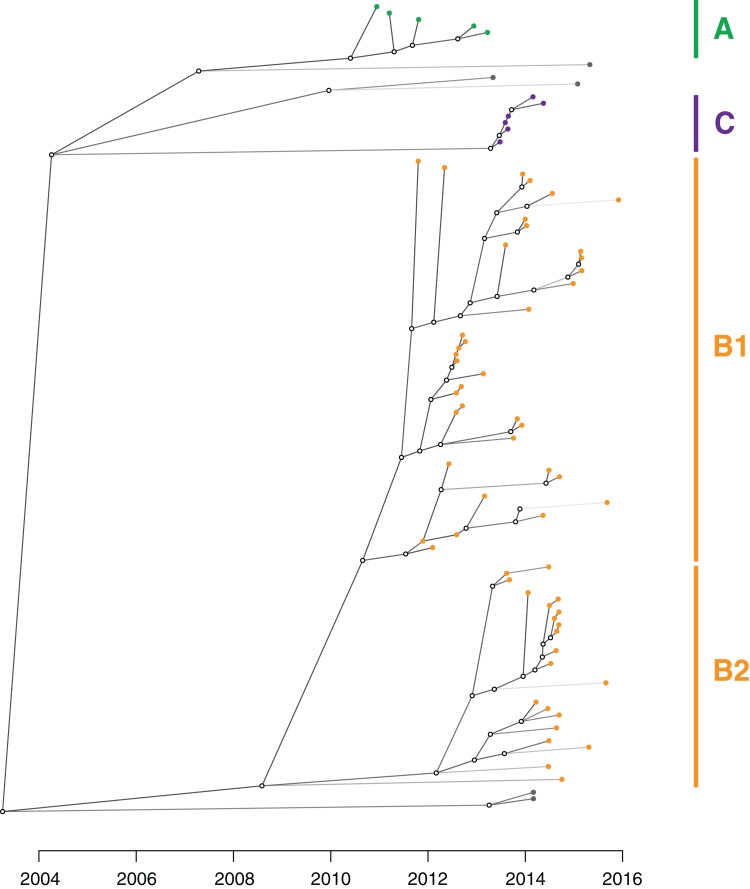
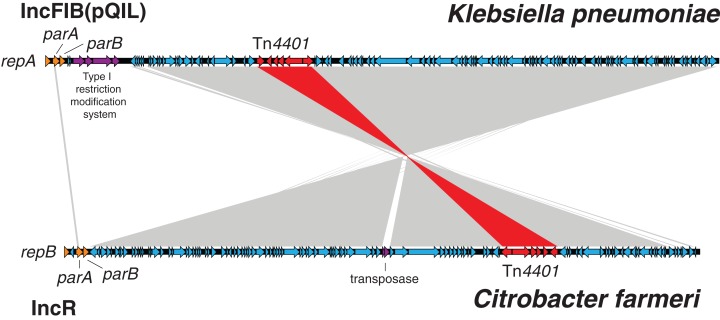
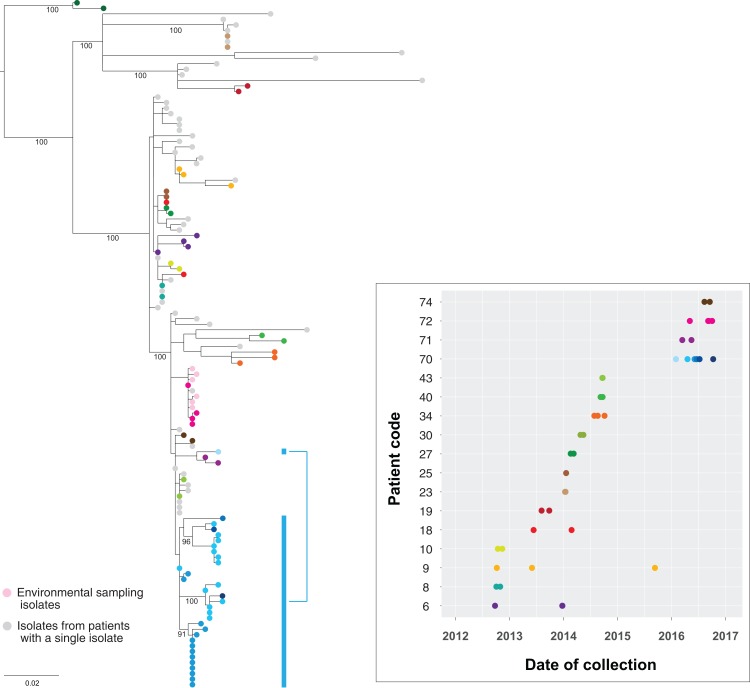
Results: Initial investigations indicated that the outbreak was widespread, with 86 KPC-producing Enterobacteriaceae isolates (K. pneumoniae 92%) identified from 35 different locations across metropolitan and rural Victoria between 2012 and 2015. Initial combined analyses of the epidemiological and genomic data resolved the outbreak into distinct nosocomial transmission networks, and identified healthcare facilities at the epicentre of KPC transmission. New cases were assigned to transmission networks in real-time, allowing focussed infection control efforts. PacBio sequencing confirmed a secondary transmission network arising from inter-species plasmid transmission. Insights from Bayesian transmission inference and analyses of within-host diversity informed the development of state-wide public health and infection control guidelines, including interventions such as an intensive approach to screening contacts following new case detection to minimise unrecognised colonisation.
Conclusion: A real-time combined epidemiological and genomic investigation proved critical to identifying and defining multiple transmission networks of KPC Enterobacteriaceae, while data from either investigation alone were inconclusive. The investigation was fundamental to informing infection control measures in real-time and the development of state-wide public health guidelines on carbapenemase-producing Enterobacteriaceae surveillance and management.
Keywords: Antimicrobial resistance; Klebsiella pneumoniae carbapenemase; Microbial genomics; Multidrug-resistant organisms; Outbreak investigation; Transmission modelling; Whole-genome sequencing.
Conflict of interest statement
Timothy P. Stinear is an Academic Editor for PeerJ.
Figures
Comment in
-
Rapid detection of carbapenemase producing Enterobacteriaceae direct from blood using real-time PCR.Pathology. 2020 Jun;52(4):499-502. doi: 10.1016/j.pathol.2020.03.008. Epub 2020 Apr 27. Pathology. 2020. PMID: 32349865 No abstract available.
References
-
- Australian Commission on Safety and Quality in Health Care . Sydney: Australian Commission on Safety and Quality in Health Care; 2013. Recommendations for the control of multi-drug resistant Gram-negatives: carbapenem resistant Enterobacteriaceae.
-
- Australian Group on Antimicrobial Resistance The evolution of carbapenemases in Enterobacteriaceae in Australia. 2014. http://www.agargroup.org/surveys http://www.agargroup.org/surveys
-
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of Computational Biology. 2012;19(5):455–477. doi: 10.1089/cmb.2012.0021. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
