

Punctuated evolution of canonical genomic aberrations in uveal melanoma
- PMID: 29317634
- PMCID: PMC5760704
- DOI: 10.1038/s41467-017-02428-w
Punctuated evolution of canonical genomic aberrations in uveal melanoma
Abstract
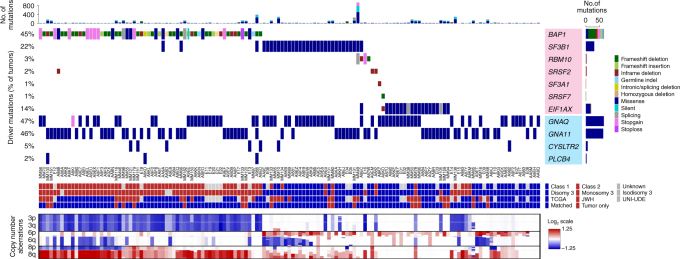
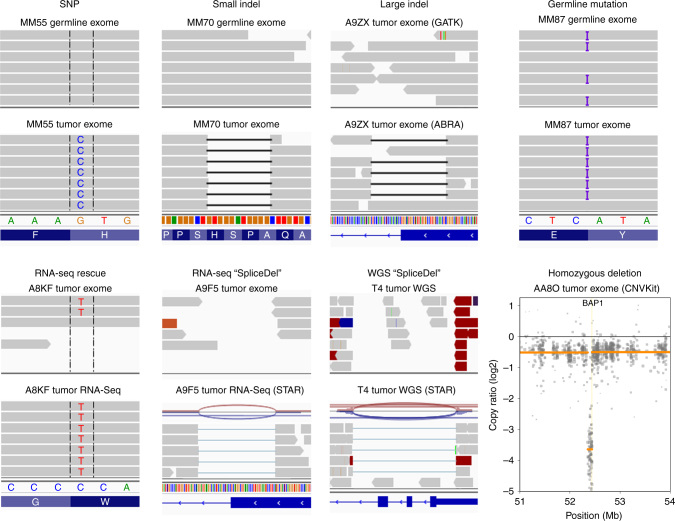
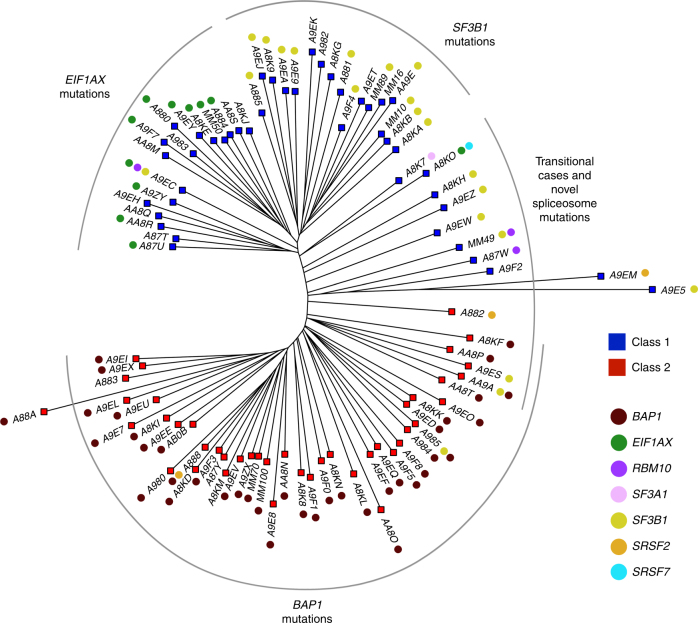
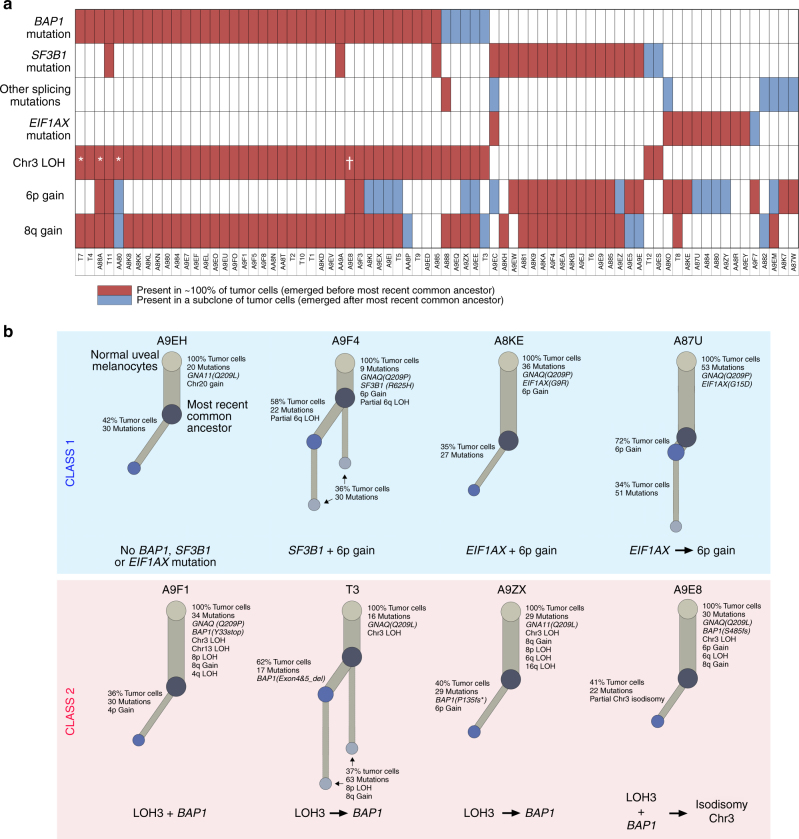
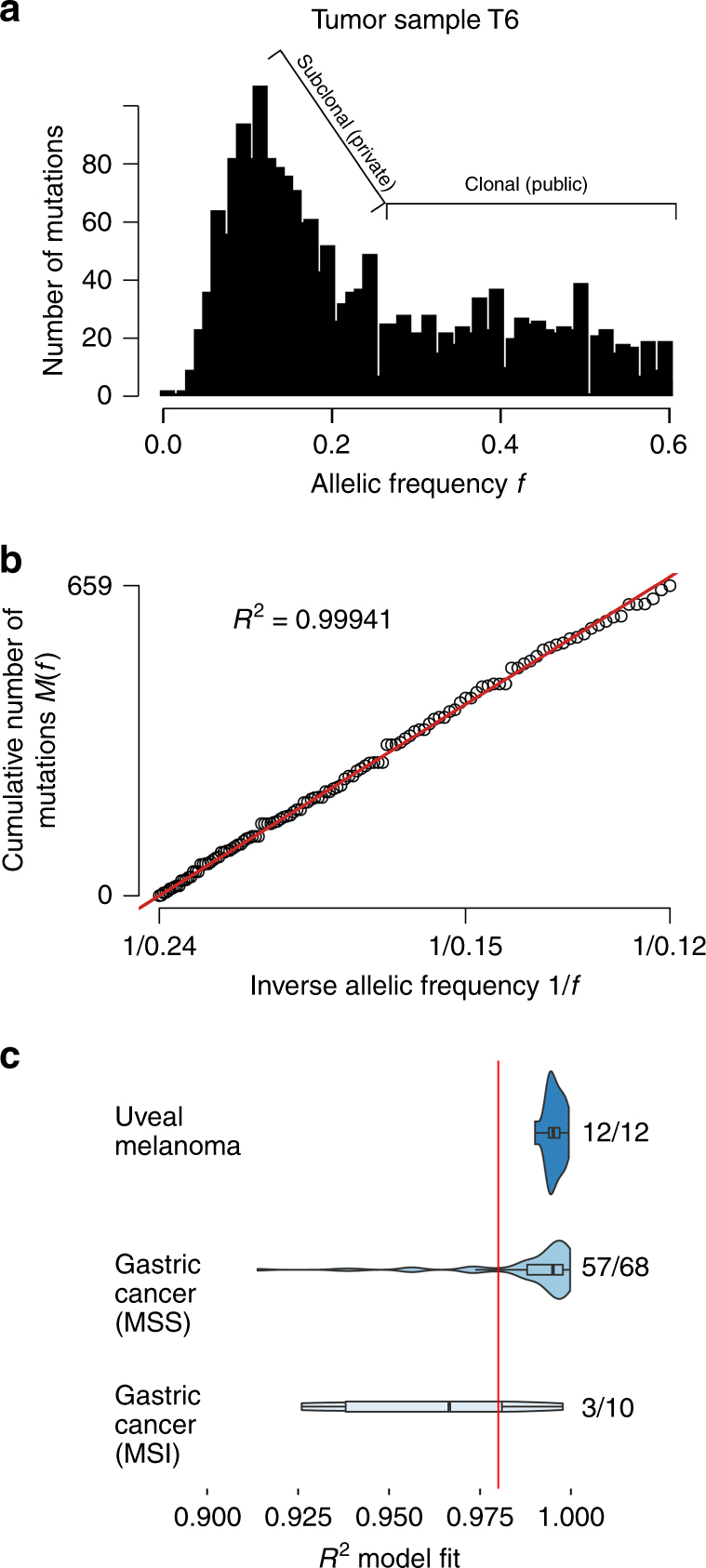
Cancer is thought to arise through the accumulation of genomic aberrations evolving under Darwinian selection. However, it remains unclear when the aberrations associated with metastasis emerge during tumor evolution. Uveal melanoma (UM) is the most common primary eye cancer and frequently leads to metastatic death, which is strongly linked to BAP1 mutations. Accordingly, UM is ideally suited for studying the clonal evolution of metastatic competence. Here we analyze sequencing data from 151 primary UM samples using a customized bioinformatic pipeline, to improve detection of BAP1 mutations and infer the clonal relationships among genomic aberrations. Strikingly, we find BAP1 mutations and other canonical genomic aberrations usually arise in an early punctuated burst, followed by neutral evolution extending to the time of clinical detection. This implies that the metastatic proclivity of UM is "set in stone" early in tumor evolution and may explain why advances in primary treatment have not improved survival.
Conflict of interest statement
Drs. Harbour and Bowcock are inventors of intellectual property discussed in this study. Dr. Harbour is a paid consultant for Castle Biosciences, licensee of this intellectual property, and he receives royalties from its commercialization. All remaining authors declare no competing financial interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
