

Loss of the Mitochondrial Fatty Acid β-Oxidation Protein Medium-Chain Acyl-Coenzyme A Dehydrogenase Disrupts Oxidative Phosphorylation Protein Complex Stability and Function
- PMID: 29317722
- PMCID: PMC5760697
- DOI: 10.1038/s41598-017-18530-4
Loss of the Mitochondrial Fatty Acid β-Oxidation Protein Medium-Chain Acyl-Coenzyme A Dehydrogenase Disrupts Oxidative Phosphorylation Protein Complex Stability and Function
Abstract
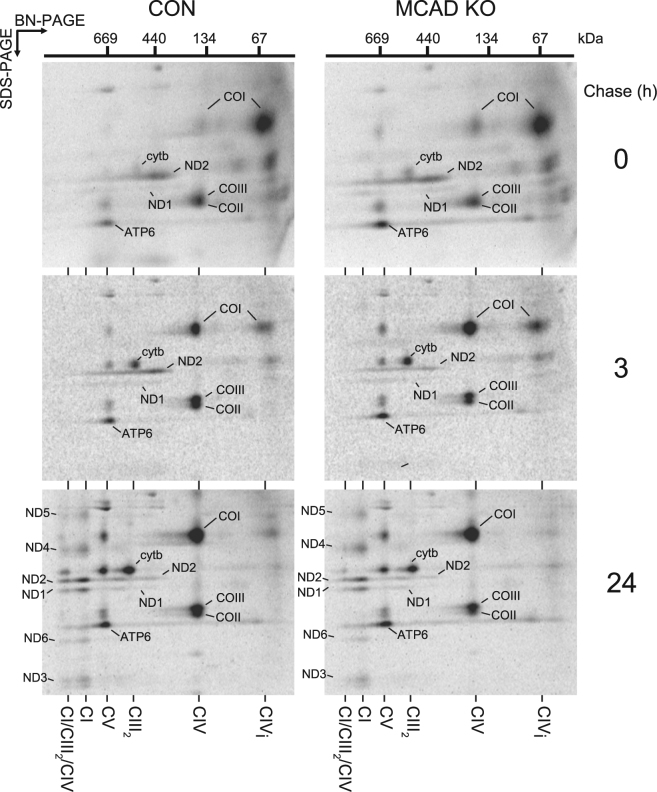
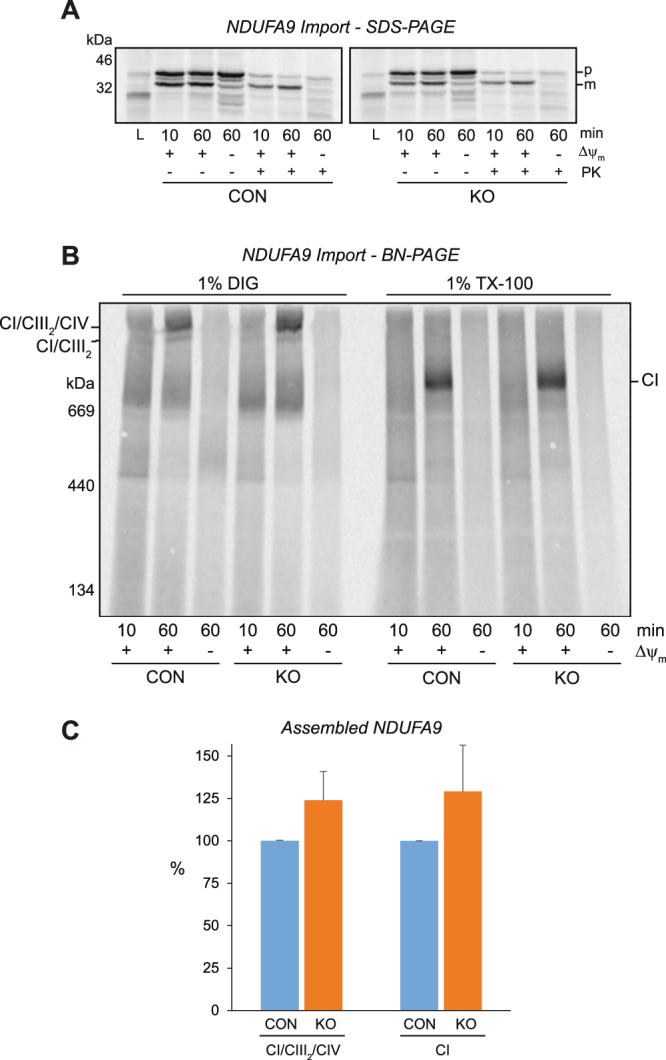
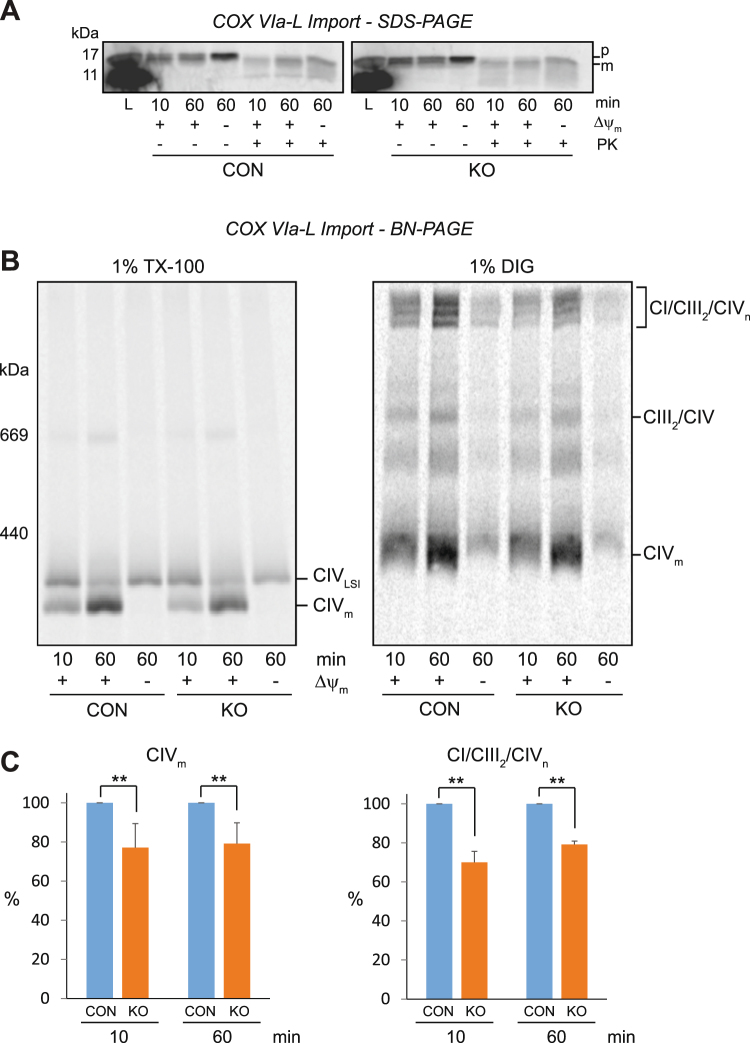
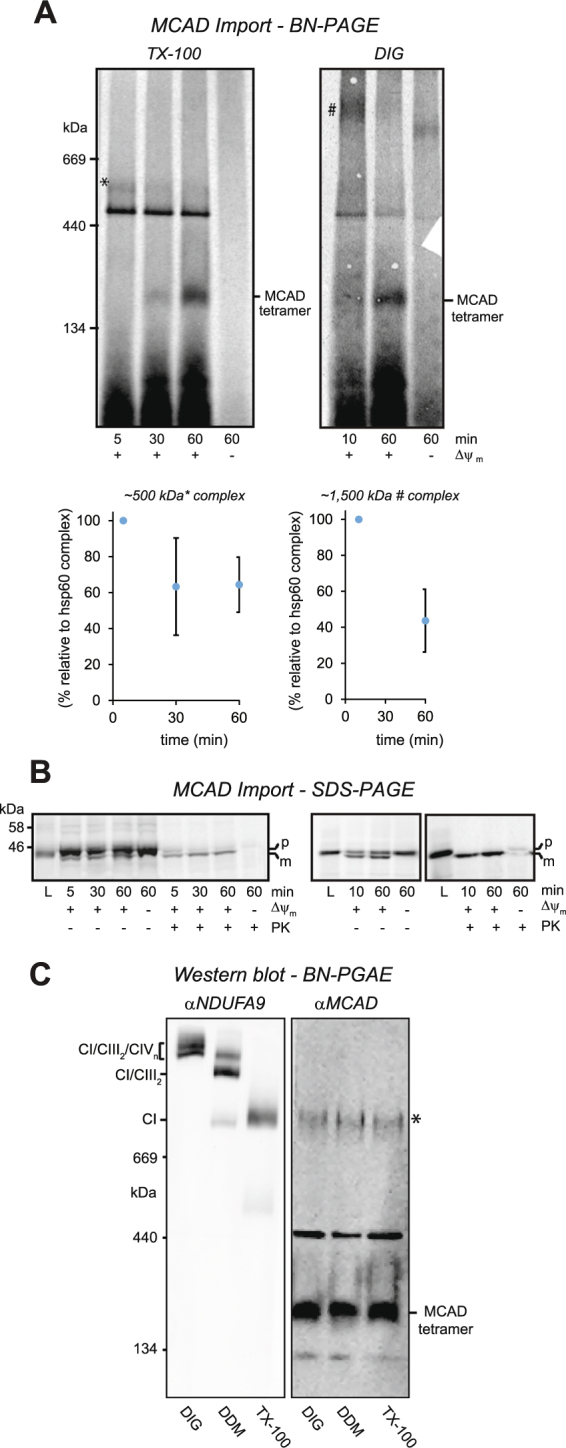
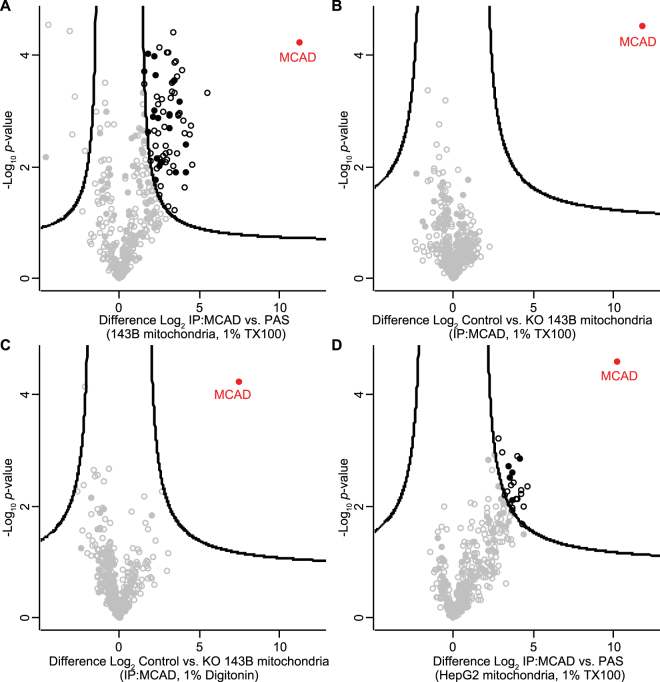
Medium-chain acyl-Coenzyme A dehydrogenase (MCAD) is involved in the initial step of mitochondrial fatty acid β-oxidation (FAO). Loss of function results in MCAD deficiency, a disorder that usually presents in childhood with hypoketotic hypoglycemia, vomiting and lethargy. While the disruption of mitochondrial fatty acid metabolism is the primary metabolic defect, secondary defects in mitochondrial oxidative phosphorylation (OXPHOS) may also contribute to disease pathogenesis. Therefore, we examined OXPHOS activity and stability in MCAD-deficient patient fibroblasts that have no detectable MCAD protein. We found a deficit in mitochondrial oxygen consumption, with reduced steady-state levels of OXPHOS complexes I, III and IV, as well as the OXPHOS supercomplex. To examine the mechanisms involved, we generated an MCAD knockout (KO) using human 143B osteosarcoma cells. These cells also exhibited defects in OXPHOS complex function and steady-state levels, as well as disrupted biogenesis of newly-translated OXPHOS subunits. Overall, our findings suggest that the loss of MCAD is associated with a reduction in steady-state OXPHOS complex levels, resulting in secondary defects in OXPHOS function which may contribute to the pathology of MCAD deficiency.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
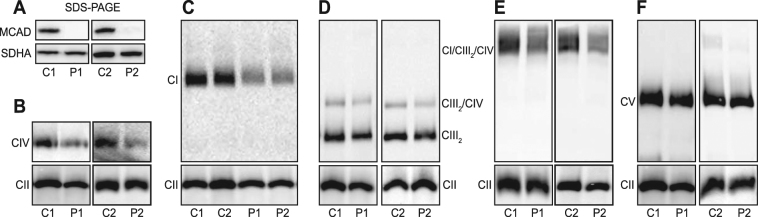
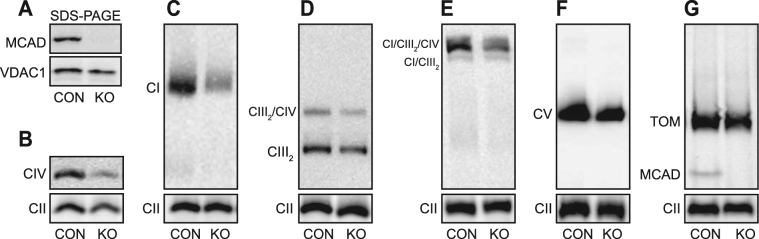
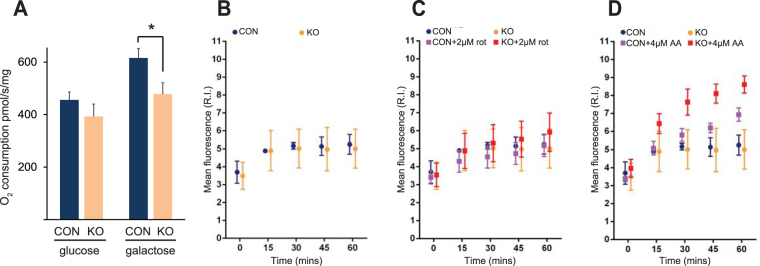
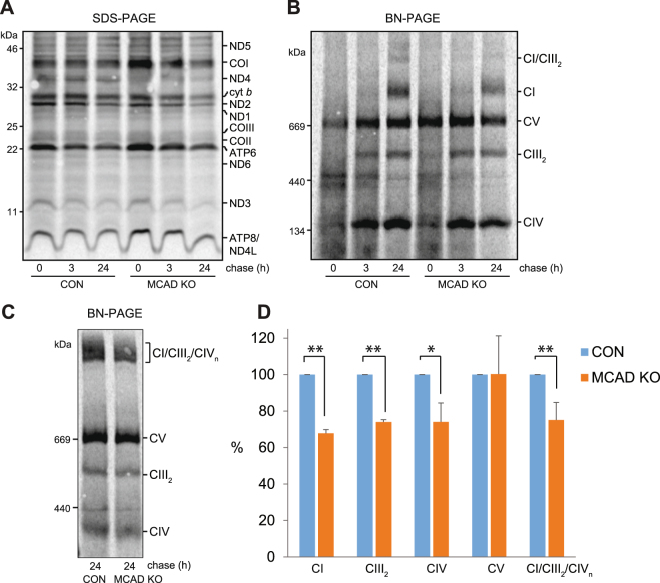
References
-
- Saijo T, Welch WJ, Tanaka K. Intramitochondrial folding and assembly of medium-chain acyl-CoA dehydrogenase (MCAD). Demonstration of impaired transfer of K304E-variant MCAD from its complex with hsp60 to the native tetramer. J. Biol. Chem. 1994;269:4401–4408. - PubMed
-
- Andresen BS, et al. Medium-chain acyl-CoA dehydrogenase (MCAD) mutations identified by MS/MS-based prospective screening of newborns differ from those observed in patients with clinical symptoms: identification and characterization of a new, prevalent mutation that results in mild MCAD deficiency. Am. J. Hum. Genet. 2001;68:1408–1418. doi: 10.1086/320602. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
