

Comparative genomic analysis of the 'pseudofungus' Hyphochytrium catenoides
- PMID: 29321239
- PMCID: PMC5795050
- DOI: 10.1098/rsob.170184
Comparative genomic analysis of the 'pseudofungus' Hyphochytrium catenoides
Abstract
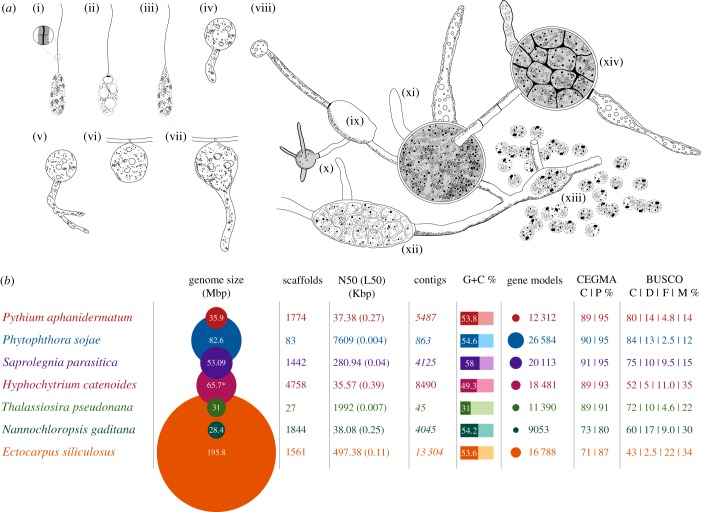
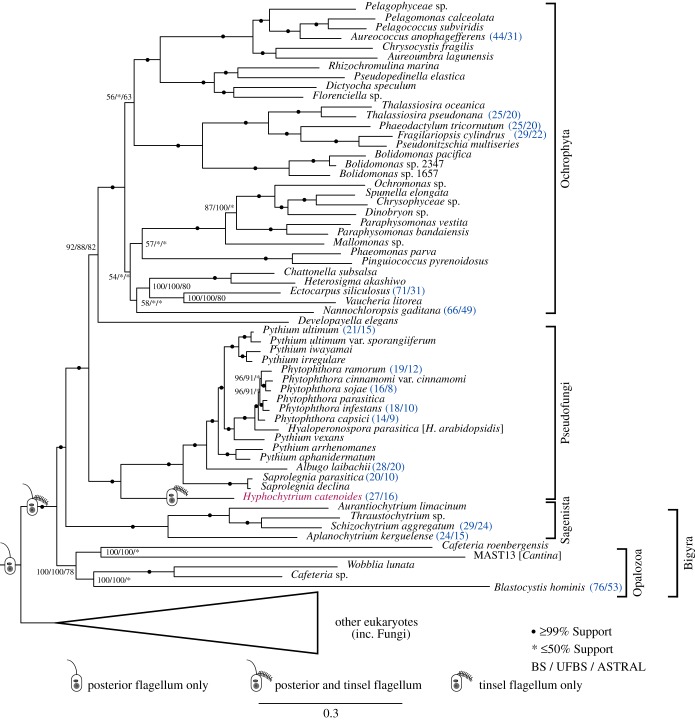
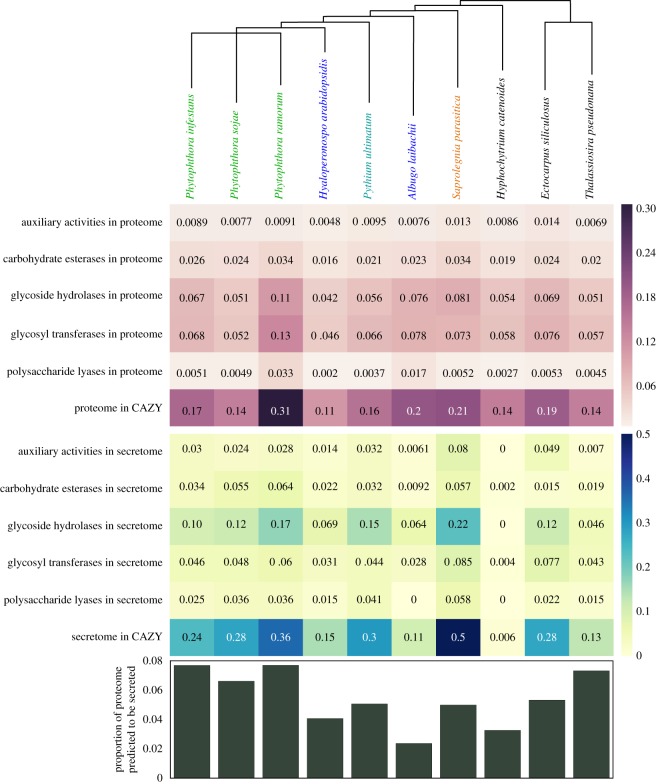
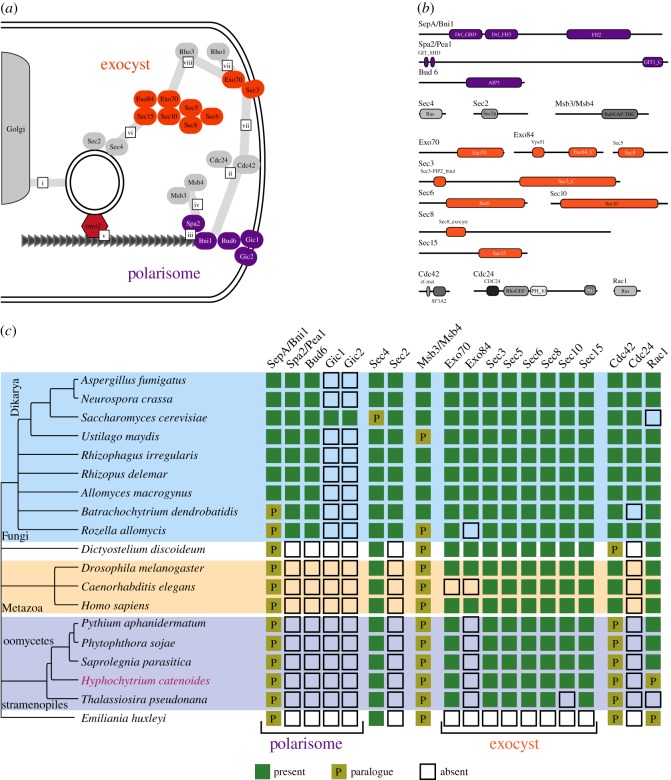
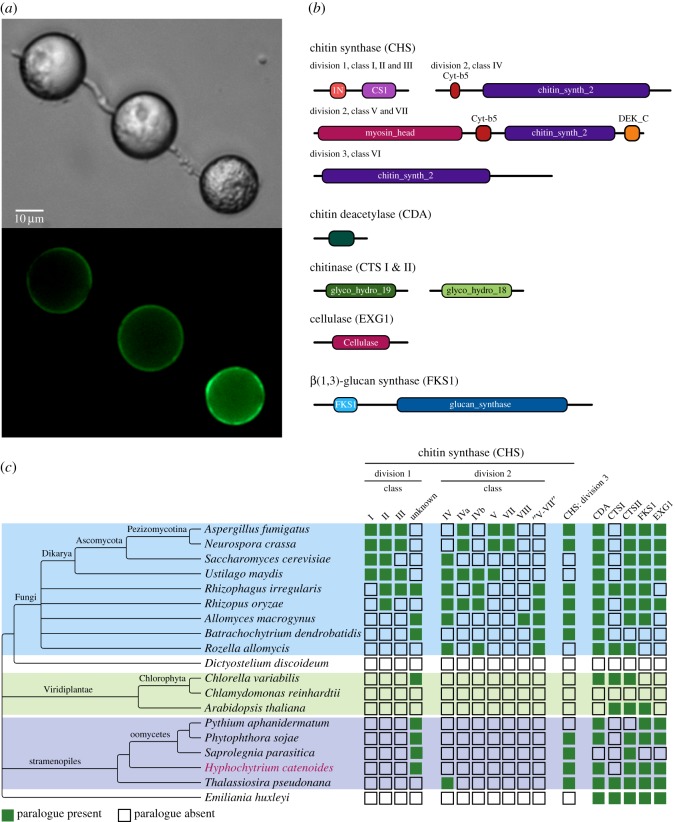
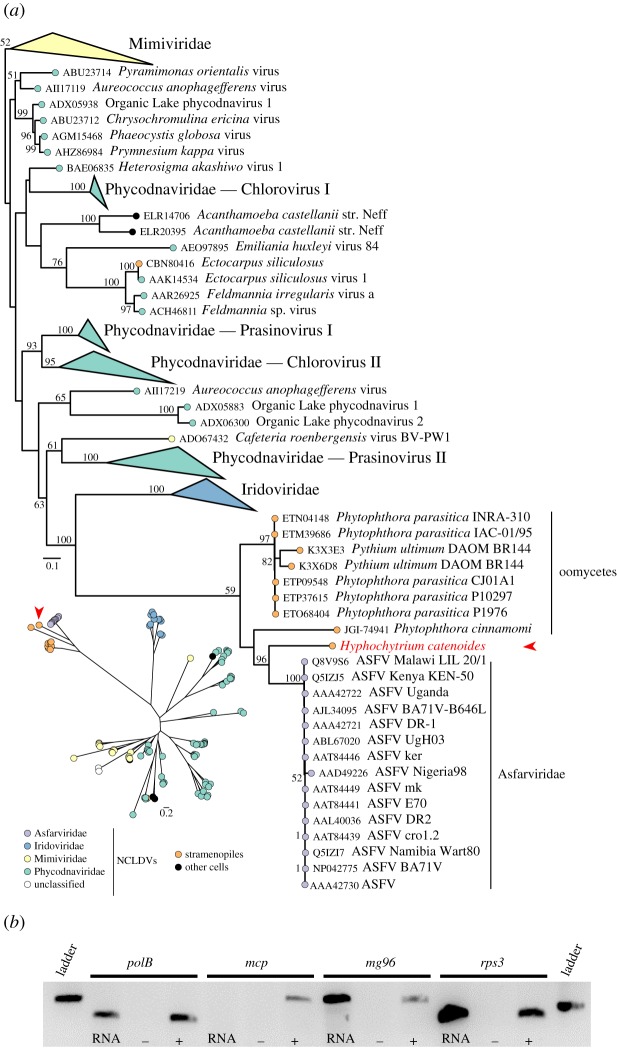
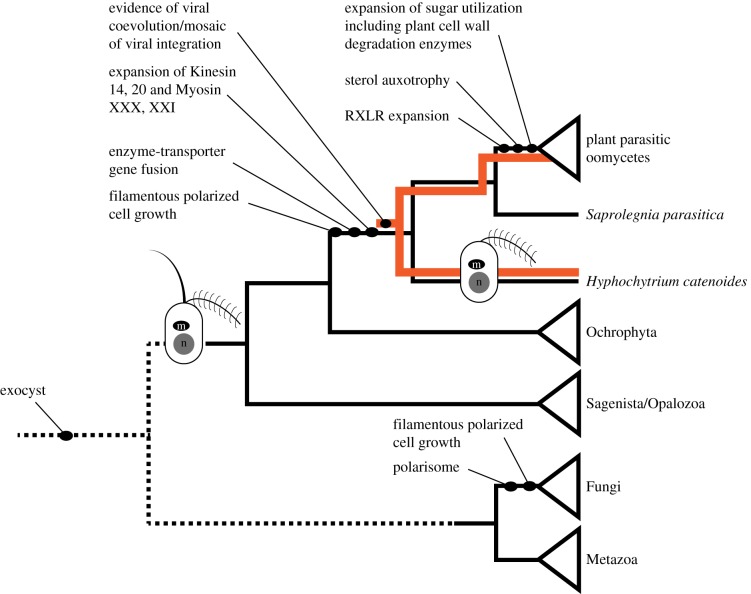
Eukaryotic microbes have three primary mechanisms for obtaining nutrients and energy: phagotrophy, photosynthesis and osmotrophy. Traits associated with the latter two functions arose independently multiple times in the eukaryotes. The Fungi successfully coupled osmotrophy with filamentous growth, and similar traits are also manifested in the Pseudofungi (oomycetes and hyphochytriomycetes). Both the Fungi and the Pseudofungi encompass a diversity of plant and animal parasites. Genome-sequencing efforts have focused on host-associated microbes (mutualistic symbionts or parasites), providing limited comparisons with free-living relatives. Here we report the first draft genome sequence of a hyphochytriomycete 'pseudofungus'; Hyphochytrium catenoides Using phylogenomic approaches, we identify genes of recent viral ancestry, with related viral derived genes also present on the genomes of oomycetes, suggesting a complex history of viral coevolution and integration across the Pseudofungi. H. catenoides has a complex life cycle involving diverse filamentous structures and a flagellated zoospore with a single anterior tinselate flagellum. We use genome comparisons, drug sensitivity analysis and high-throughput culture arrays to investigate the ancestry of oomycete/pseudofungal characteristics, demonstrating that many of the genetic features associated with parasitic traits evolved specifically within the oomycete radiation. Comparative genomics also identified differences in the repertoire of genes associated with filamentous growth between the Fungi and the Pseudofungi, including differences in vesicle trafficking systems, cell-wall synthesis pathways and motor protein repertoire, demonstrating that unique cellular systems underpinned the convergent evolution of filamentous osmotrophic growth in these two eukaryotic groups.
Keywords: large DNA virus; oomycete parasitic traits; polarized filamentous growth; secondary plastid endosymbiosis.
© 2018 The Authors.
Conflict of interest statement
We declare we have no competing interests.
Figures

References
-
- Patterson DJ. 1989. Stamenopiles: chromophytes from a protistan perspective. In The chromophyte algae, problems and perspectives. (eds Green JC, Leadbeater BSC, Diver WL), pp. 357–379. Oxford, UK: Clarendon.
-
- Cavalier-Smith T. 1986. The kingdom Chromista: origin and systematics. In Progress in phycological research (eds Round FE, Chapman DJ), pp. 309–347. Bristol, UK: Biopress Ltd.
-
- Cavalier-Smith T, Chao EE. 2006. Phylogeny and megasystematics of phagotrophic heterokonts (kingdom Chromista). J. Mol. Evol. 62, 388–420. (doi:10.1007/s00239-004-0353-8) - DOI - PubMed
-
- Cavalier-Smith T. 1987. The origin of fungi and pseudofungi. In Evolutionary biology of the fungi (British mycological society symposia). (ed. Rayer ADM.), pp. 339–353. Cambridge, UK: Cambridge University Press.
-
- Burki F, Kaplan M, Tikhonenkov DV, Zlatogursky V, Minh BQ, Radaykina LV, Smirnov A, Mylnikov AP, Keeling PJ. 2016. Untangling the early diversification of eukaryotes: a phylogenomic study of the evolutionary origins of Centrohelida, Haptophyta and Cryptista. Proc. R. Soc. B 283, 20152802 (doi:10.1098/rspb.2015.2802) - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources