

Integrative genomic and transcriptomic analysis of leiomyosarcoma
- PMID: 29321523
- PMCID: PMC5762758
- DOI: 10.1038/s41467-017-02602-0
Integrative genomic and transcriptomic analysis of leiomyosarcoma
Abstract
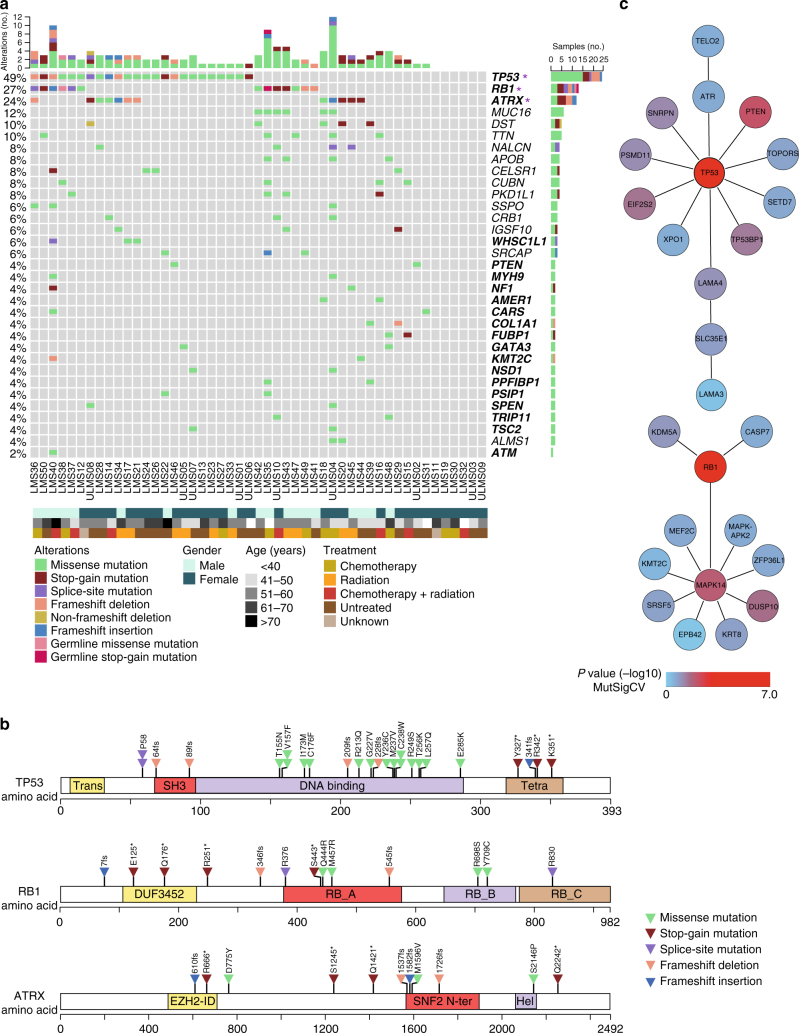
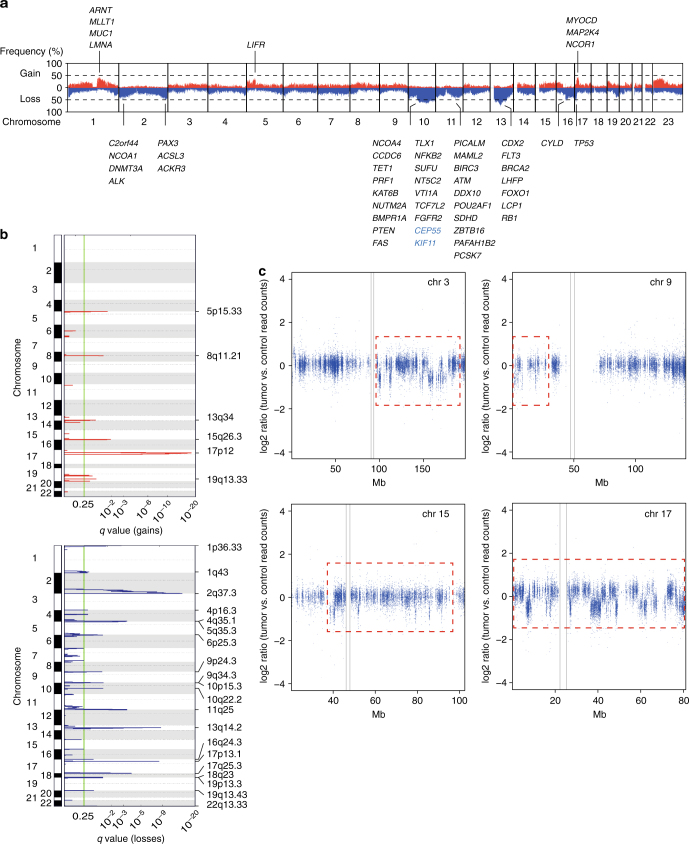
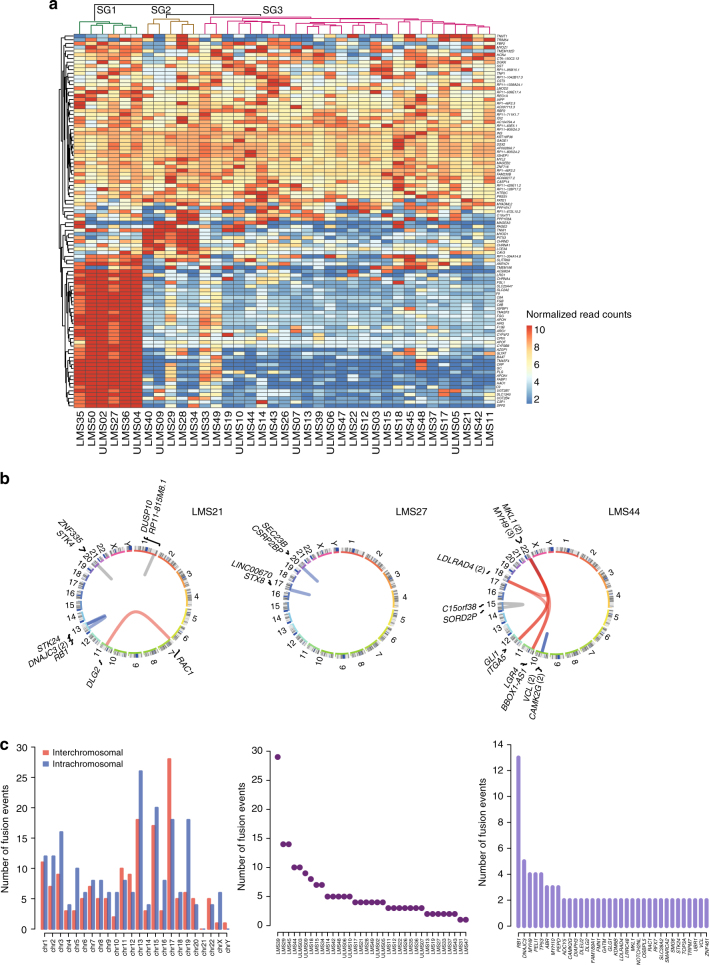
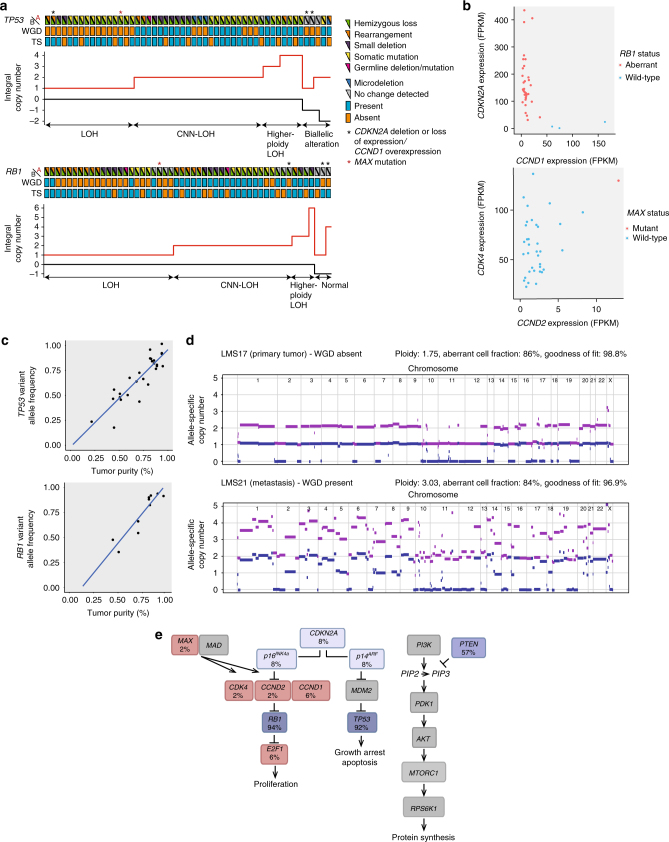
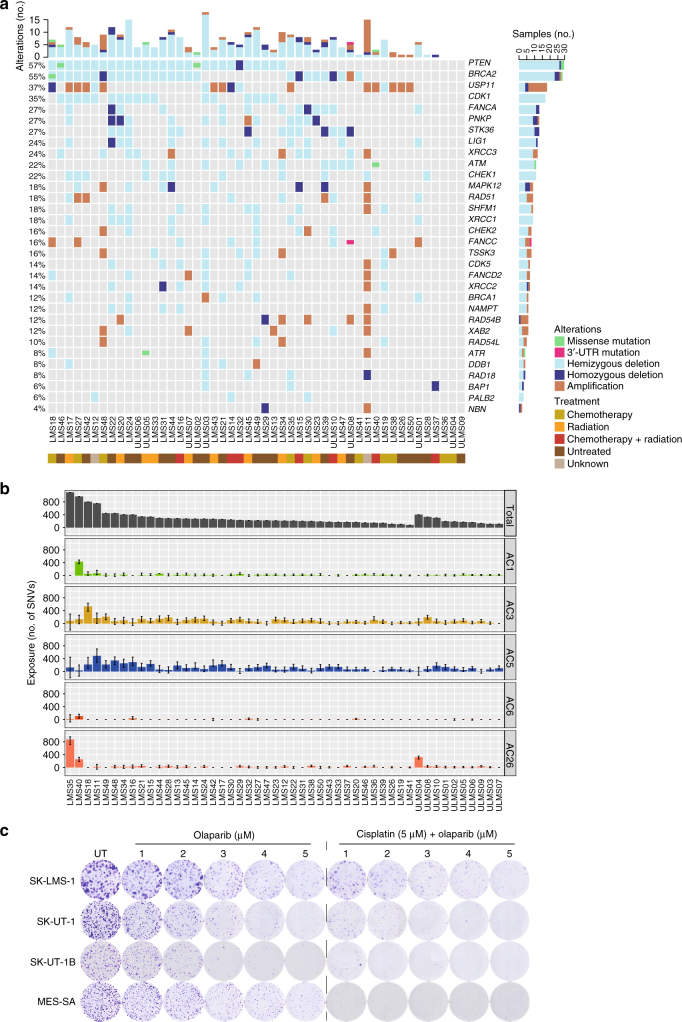
Leiomyosarcoma (LMS) is an aggressive mesenchymal malignancy with few therapeutic options. The mechanisms underlying LMS development, including clinically actionable genetic vulnerabilities, are largely unknown. Here we show, using whole-exome and transcriptome sequencing, that LMS tumors are characterized by substantial mutational heterogeneity, near-universal inactivation of TP53 and RB1, widespread DNA copy number alterations including chromothripsis, and frequent whole-genome duplication. Furthermore, we detect alternative telomere lengthening in 78% of cases and identify recurrent alterations in telomere maintenance genes such as ATRX, RBL2, and SP100, providing insight into the genetic basis of this mechanism. Finally, most tumors display hallmarks of "BRCAness", including alterations in homologous recombination DNA repair genes, multiple structural rearrangements, and enrichment of specific mutational signatures, and cultured LMS cells are sensitive towards olaparib and cisplatin. This comprehensive study of LMS genomics has uncovered key biological features that may inform future experimental research and enable the design of novel therapies.
Conflict of interest statement
The authors declare no competing financial interests.
Figures


References
-
- Coindre JM, et al. Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: a study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer. 2001;91:1914–1926. doi: 10.1002/1097-0142(20010515)91:10<1914::AID-CNCR1214>3.0.CO;2-3. - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
