

Clinical and experimental evidence suggest a link between KIF7 and C5orf42-related ciliopathies through Sonic Hedgehog signaling
- PMID: 29321670
- PMCID: PMC5839020
- DOI: 10.1038/s41431-017-0019-9
Clinical and experimental evidence suggest a link between KIF7 and C5orf42-related ciliopathies through Sonic Hedgehog signaling
Abstract
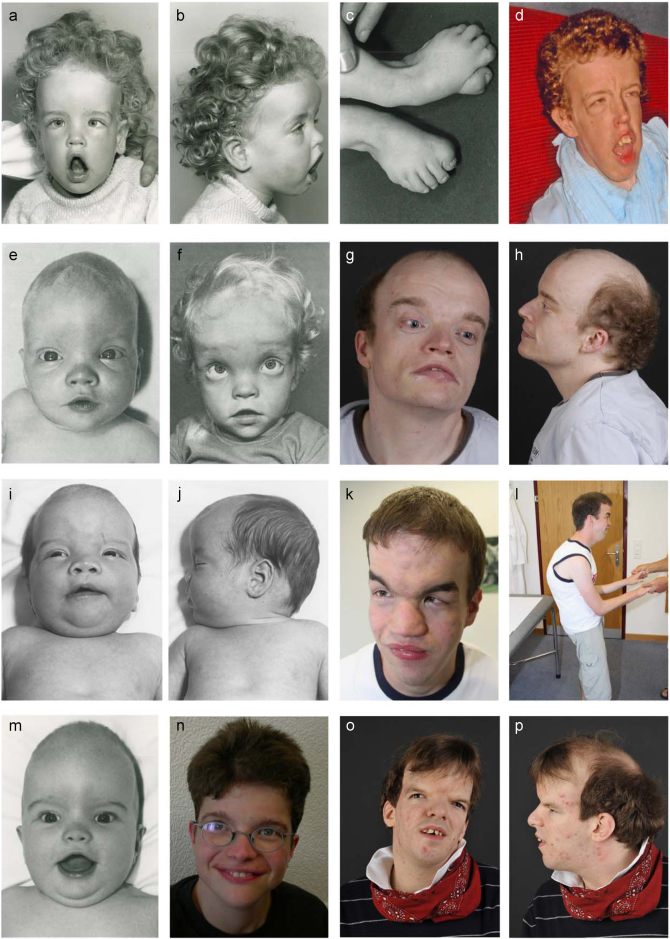
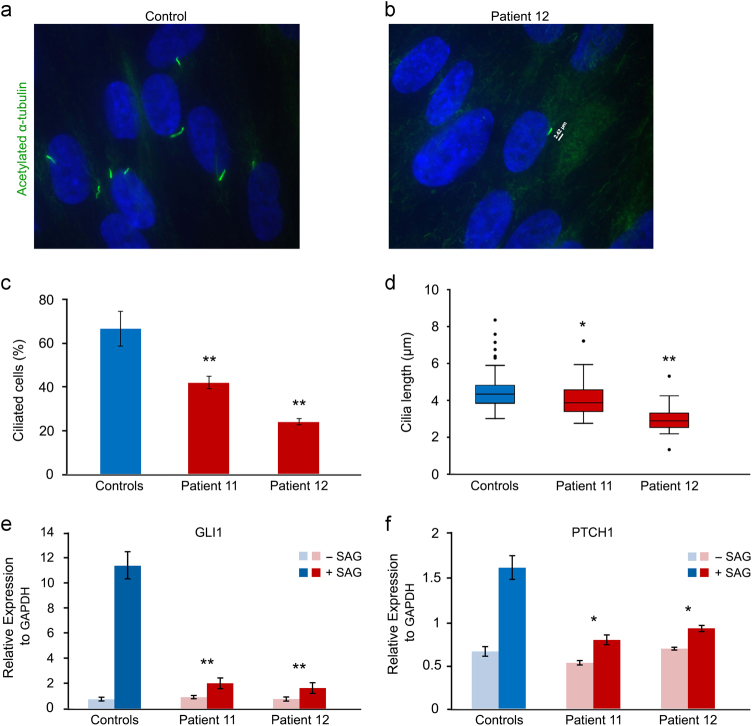
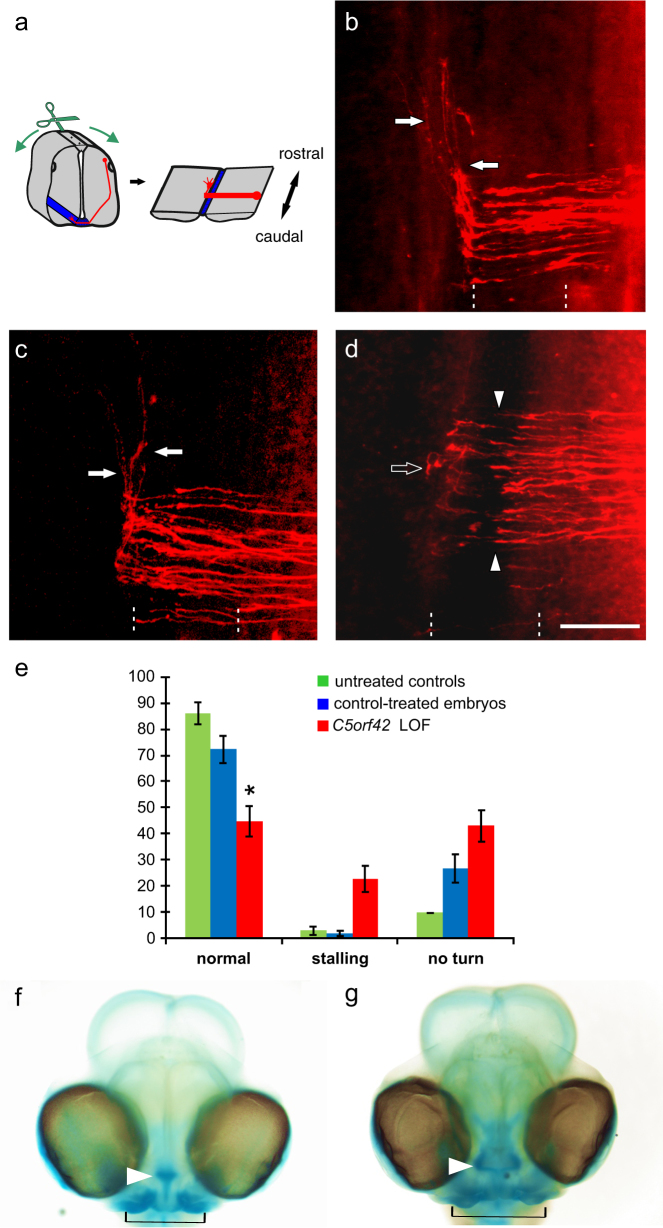
Acrocallosal syndrome (ACLS) is an autosomal recessive neurodevelopmental disorder caused by KIF7 defects and belongs to the heterogeneous group of ciliopathies related to Joubert syndrome (JBTS). While ACLS is characterized by macrocephaly, prominent forehead, depressed nasal bridge, and hypertelorism, facial dysmorphism has not been emphasized in JBTS cohorts with molecular diagnosis. To evaluate the specificity and etiology of ACLS craniofacial features, we performed whole exome or targeted Sanger sequencing in patients with the aforementioned overlapping craniofacial appearance but variable additional ciliopathy features followed by functional studies. We found (likely) pathogenic variants of KIF7 in 5 out of 9 families, including the original ACLS patients, and delineated 1000 to 4000-year-old Swiss founder alleles. Three of the remaining families had (likely) pathogenic variants in the JBTS gene C5orf42, and one patient had a novel de novo frameshift variant in SHH known to cause autosomal dominant holoprosencephaly. In accordance with the patients' craniofacial anomalies, we showed facial midline widening after silencing of C5orf42 in chicken embryos. We further supported the link between KIF7, SHH, and C5orf42 by demonstrating abnormal primary cilia and diminished response to a SHH agonist in fibroblasts of C5orf42-mutated patients, as well as axonal pathfinding errors in C5orf42-silenced chicken embryos similar to those observed after perturbation of Shh signaling. Our findings, therefore, suggest that beside the neurodevelopmental features, macrocephaly and facial widening are likely more general signs of disturbed SHH signaling. Nevertheless, long-term follow-up revealed that C5orf42-mutated patients showed catch-up development and fainting of facial features contrary to KIF7-mutated patients.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

References
-
- Schinzel A. Postaxial polydactyly, hallux duplication, absence of the corpus callosum, macrencephaly and severe mental retardation: a new syndrome? Helv Paediatr Acta. 1979;34:141–6. - PubMed
-
- Parisi M, Glass I. Joubert syndrome and related disorders. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mefford HC, Stephens K, Amemiya A, Ledbetter N (eds). SourceGeneReviews® [Internet]. University of Washington: Seattle, USA, 1993-2013. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
