

Epithelial-mesenchymal transition leads to NK cell-mediated metastasis-specific immunosurveillance in lung cancer
- PMID: 29324443
- PMCID: PMC5873856
- DOI: 10.1172/JCI97611
Epithelial-mesenchymal transition leads to NK cell-mediated metastasis-specific immunosurveillance in lung cancer
Abstract
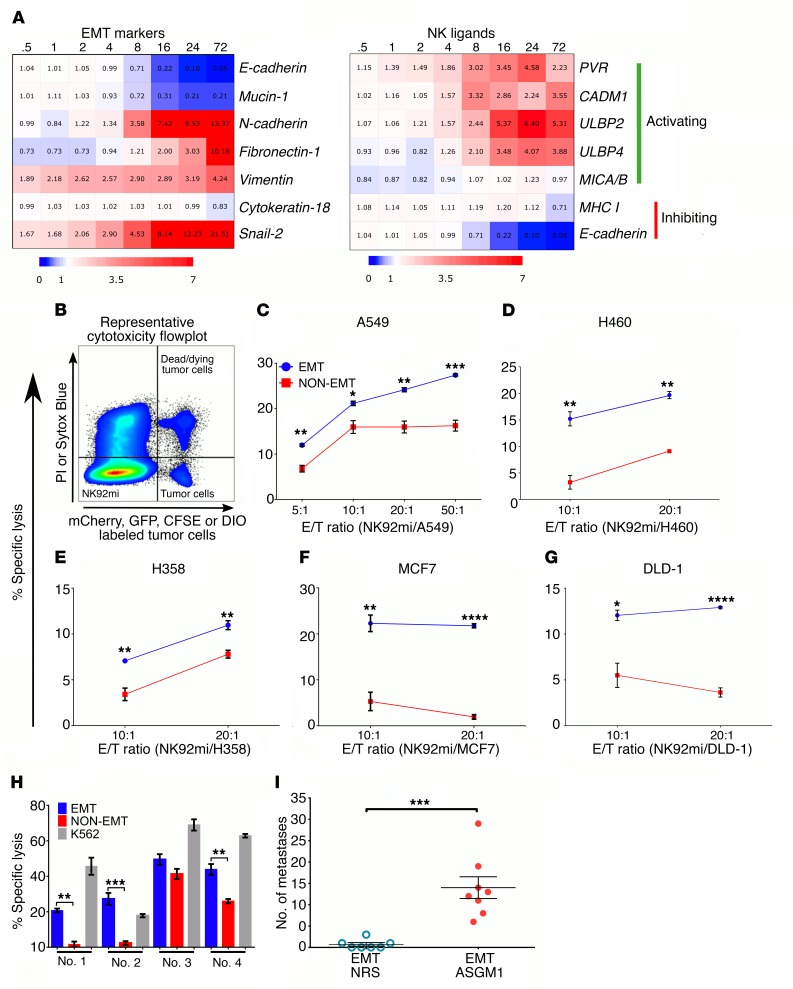
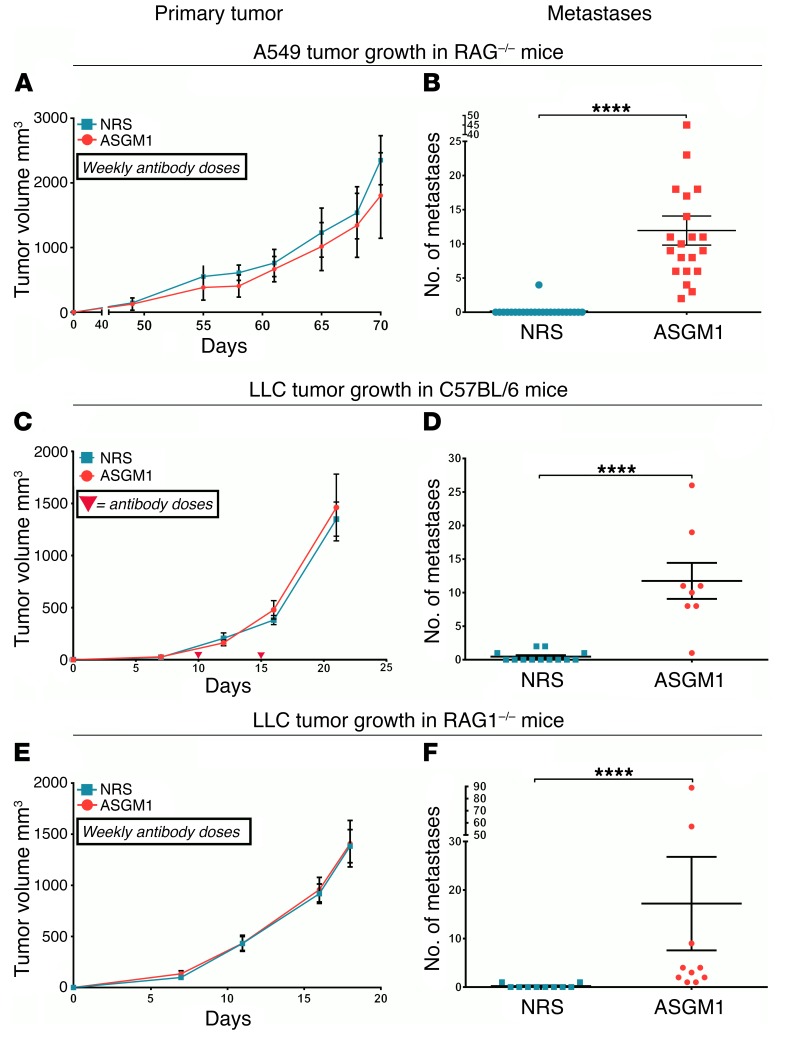
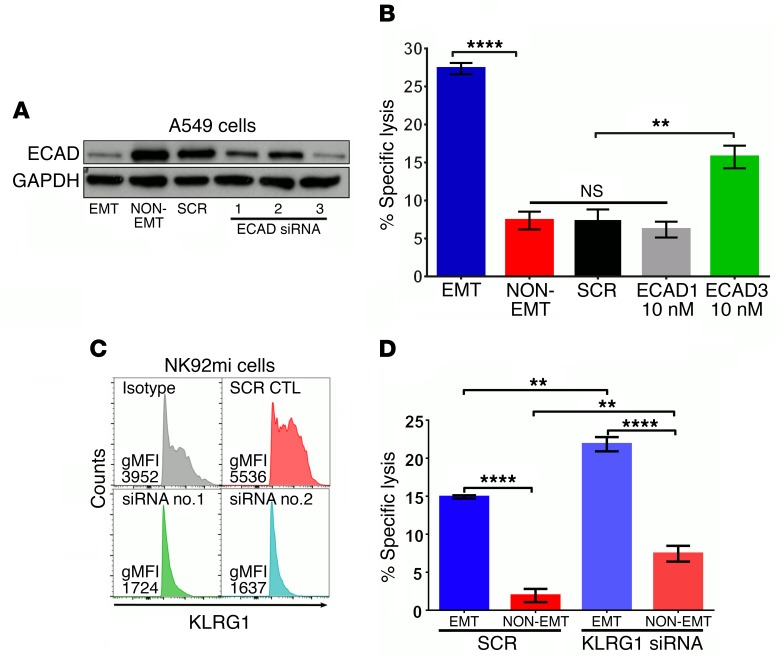
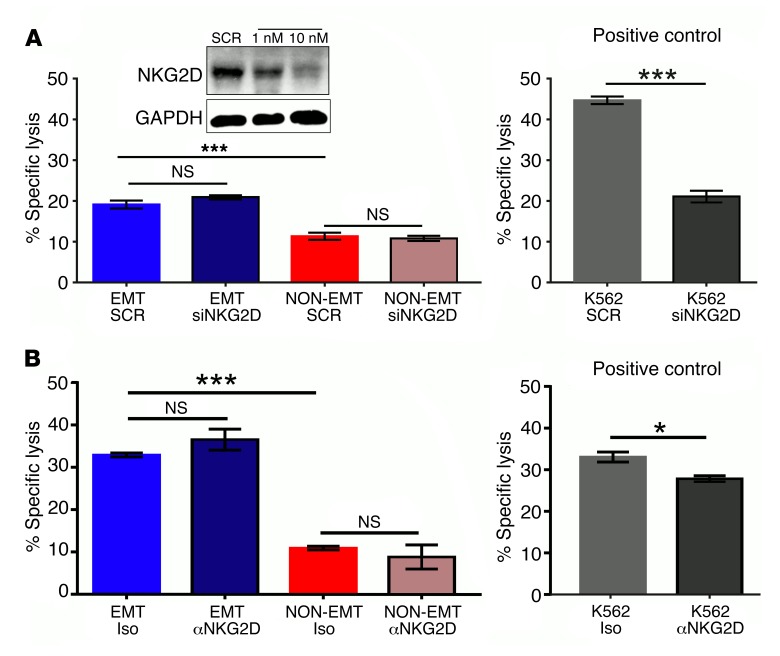
During epithelial-mesenchymal transition (EMT) epithelial cancer cells transdifferentiate into highly motile, invasive, mesenchymal-like cells, giving rise to disseminating tumor cells. Few of these disseminated cells successfully metastasize. Immune cells and inflammation in the tumor microenvironment were shown to drive EMT, but few studies investigated the consequences of EMT for tumor immunosurveillance. In addition to initiating metastasis, we demonstrate that EMT confers increased susceptibility to natural killer (NK) cells and contributes, in part, to the inefficiency of the metastatic process. Depletion of NK cells allowed spontaneous metastasis without affecting primary tumor growth. EMT-induced modulation of E-cadherin and cell adhesion molecule 1 (CADM1) mediated increased susceptibility to NK cytotoxicity. Higher CADM1 expression correlates with improved patient survival in 2 lung and 1 breast adenocarcinoma patient cohorts and decreased metastasis. Our observations reveal a novel NK-mediated, metastasis-specific immunosurveillance in lung cancer and present a window of opportunity for preventing metastasis by boosting NK cell activity.
Keywords: Immunology; Immunotherapy; Lung cancer; NK cells; Oncology.
Conflict of interest statement
Figures
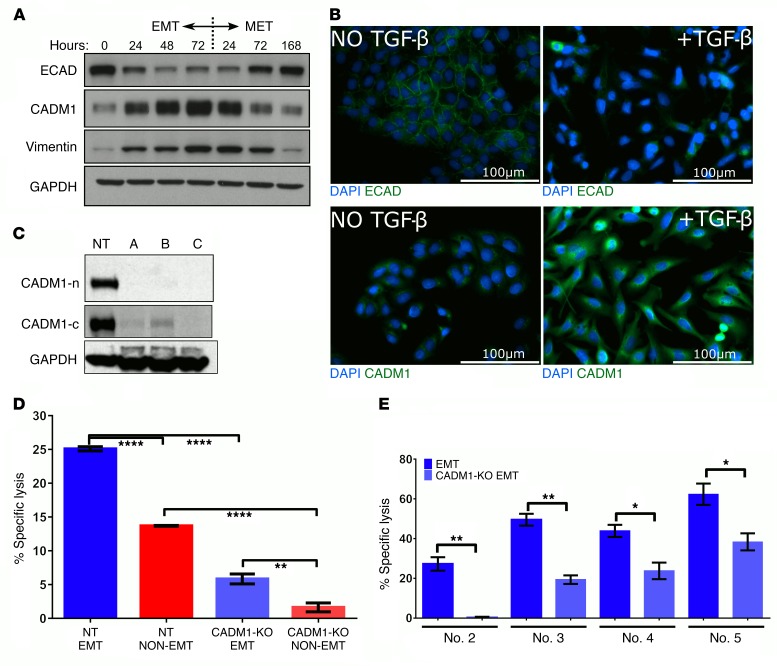
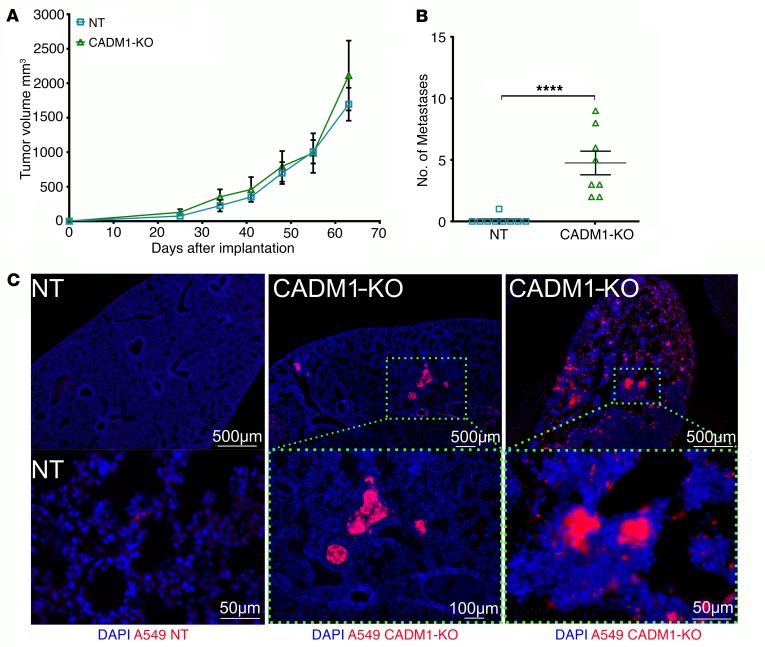
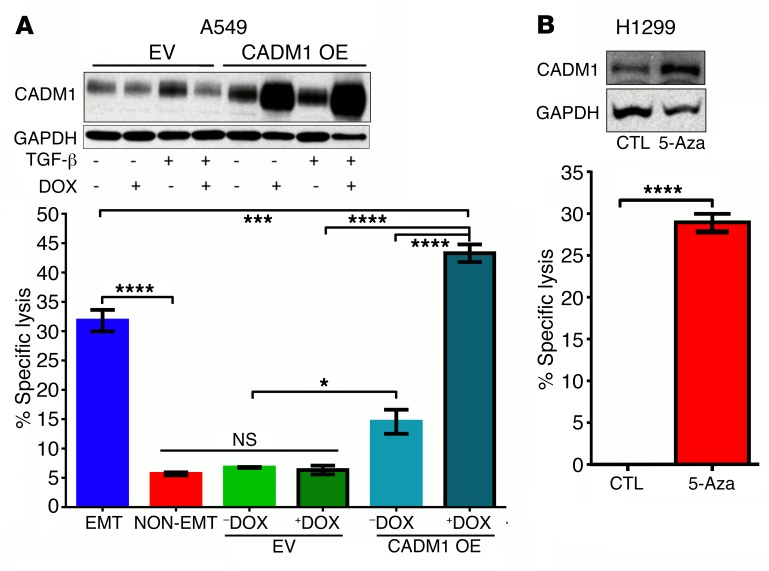
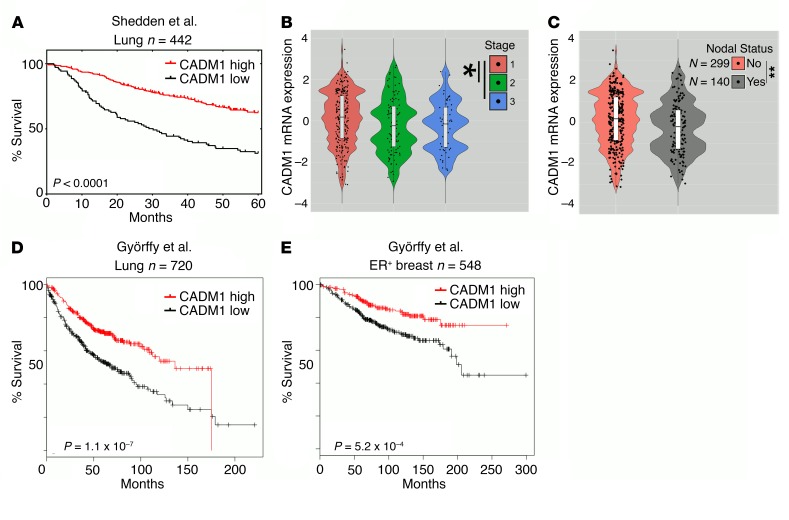
Comment in
-
Epithelial-mesenchymal transition-induced metastasis could be a bait for natural killer cells.J Thorac Dis. 2018 Sep;10(Suppl 26):S3143-S3146. doi: 10.21037/jtd.2018.08.19. J Thorac Dis. 2018. PMID: 30370099 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
