

Repeat expansion diseases
- PMID: 29325606
- PMCID: PMC6485936
- DOI: 10.1016/B978-0-444-63233-3.00009-9
Repeat expansion diseases
Abstract
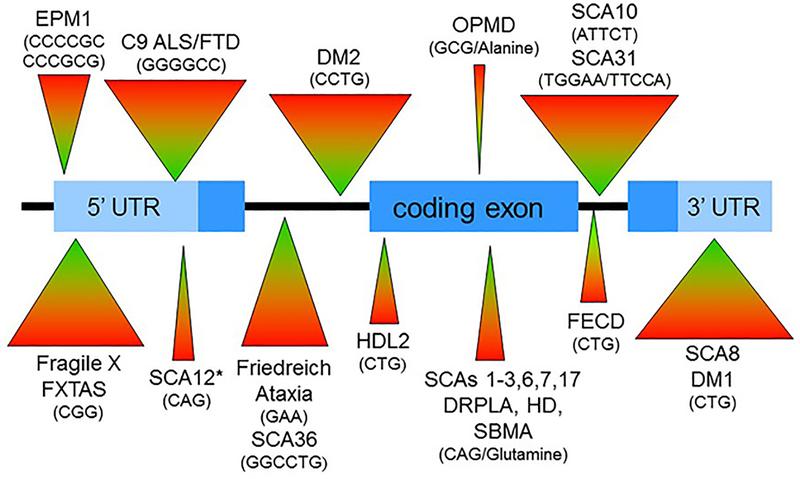
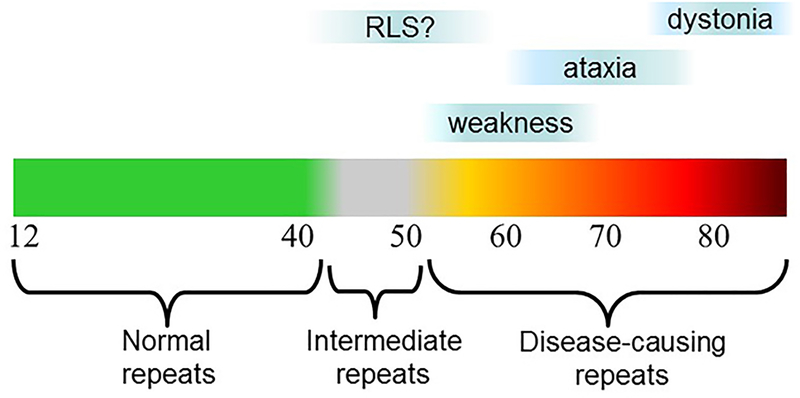
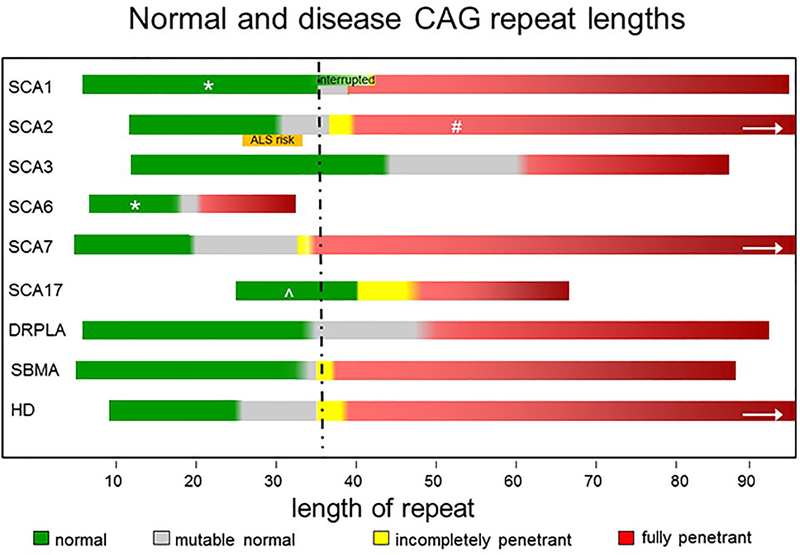
More than 40 diseases, most of which primarily affect the nervous system, are caused by expansions of simple sequence repeats dispersed throughout the human genome. Expanded trinucleotide repeat diseases were discovered first and remain the most frequent. More recently tetra-, penta-, hexa-, and even dodeca-nucleotide repeat expansions have been identified as the cause of human disease, including some of the most common genetic disorders seen by neurologists. Repeat expansion diseases include both causes of myotonic dystrophy (DM1 and DM2), the most common genetic cause of amyotrophic lateral sclerosis/frontotemporal dementia (C9ORF72), Huntington disease, and eight other polyglutamine disorders, including the most common forms of dominantly inherited ataxia, the most common recessive ataxia (Friedreich ataxia), and the most common heritable mental retardation (fragile X syndrome). Here I review distinctive features of this group of diseases that stem from the unusual, dynamic nature of the underlying mutations. These features include marked clinical heterogeneity and the phenomenon of clinical anticipation. I then discuss the diverse molecular mechanisms driving disease pathogenesis, which vary depending on the repeat sequence, size, and location within the disease gene, and whether the repeat is translated into protein. I conclude with a brief clinical and genetic description of individual repeat expansion diseases that are most relevant to neurologists.
Keywords: C9ORF72; anticipation; expanded repeats; myotonic dystrophy; polyglutamine diseases; repeat instability; spinocerebellar ataxia; trinucleotide.
Copyright © 2018 Elsevier B.V. All rights reserved.
Figures
References
-
- Anderson DG, Walker RH, Connor M, et al. (2017). A systematic review of the Huntington disease-like 2 phenotype. J Huntingtons Dis 6:37–46. - PubMed
-
- Andrew SE, Goldberg YP, Kremer B, et al. (1993). The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nat Genet 4:398–403. - PubMed
-
- Anheim M, Mariani LL, Calvas P, et al. (2012). Exonic deletions of FXN and early-onset Friedreich ataxia. Arch Neurol 69:912–6. - PubMed
-
- Anvret M, Ahlberg G, Grandell U, et al. (1993). Larger expansions of the CTG repeat in muscle compared to lymphocytes from patients with myotonic dystrophy. Hum Mol Genet 2:1397–400. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
