

Systematic quantitative analysis of H2A and H2B variants by targeted proteomics
- PMID: 29329550
- PMCID: PMC5767011
- DOI: 10.1186/s13072-017-0172-y
Systematic quantitative analysis of H2A and H2B variants by targeted proteomics
Abstract
Background: Histones organize DNA into chromatin through a variety of processes. Among them, a vast diversity of histone variants can be incorporated into chromatin and finely modulate its organization and functionality. Classically, the study of histone variants has largely relied on antibody-based assays. However, antibodies have a limited efficiency to discriminate between highly similar histone variants.
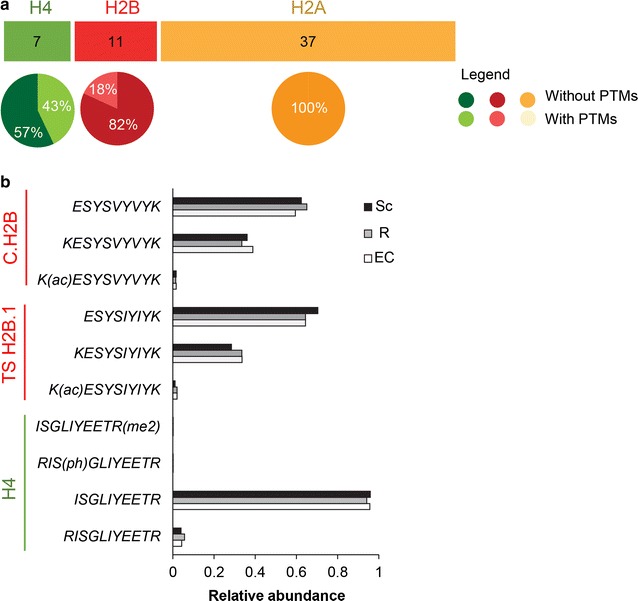
Results: In this study, we established a mass spectrometry-based analysis to address this challenge. We developed a targeted proteomics method, using selected reaction monitoring or parallel reaction monitoring, to quantify a maximum number of histone variants in a single multiplexed assay, even when histones are present in a crude extract. This strategy was developed on H2A and H2B variants, using 55 peptides corresponding to 25 different histone sequences, among which a few differ by a single amino acid. The methodology was then applied to mouse testis extracts in which almost all histone variants are expressed. It confirmed the abundance profiles of several testis-specific histones during successive stages of spermatogenesis and the existence of predicted H2A.L.1 isoforms. This methodology was also used to explore the over-expression pattern of H2A.L.1 isoforms in a mouse model of male infertility.
Conclusions: Our results demonstrate that targeted proteomics is a powerful method to quantify highly similar histone variants and isoforms. The developed method can be easily transposed to the study of human histone variants, whose abundance can be deregulated in various diseases.
Keywords: Chromatin; Histone variants; PRM; Proteomics; SRM; Spermatogenesis; Targeted proteomics.
Figures






References
Publication types
MeSH terms
Substances
Grants and funding
- ANR-11-PDOC-0011/Agence Nationale de la Recherche/International
- ANR-12-JSV2-0005-01/Agence Nationale de la Recherche/International
- ANR-10-LABX-49-01/Agence Nationale de la Recherche/International
- ANR-14-CE19-0014-01/Agence Nationale de la Recherche (FR)/International
- ANR-10-INBS-08/Agence Nationale de la Recherche (FR)/International
LinkOut - more resources
Full Text Sources
Other Literature Sources
