

Development of an Efficient Genome Editing Tool in Bacillus licheniformis Using CRISPR-Cas9 Nickase
- PMID: 29330178
- PMCID: PMC5835740
- DOI: 10.1128/AEM.02608-17
Development of an Efficient Genome Editing Tool in Bacillus licheniformis Using CRISPR-Cas9 Nickase
Abstract
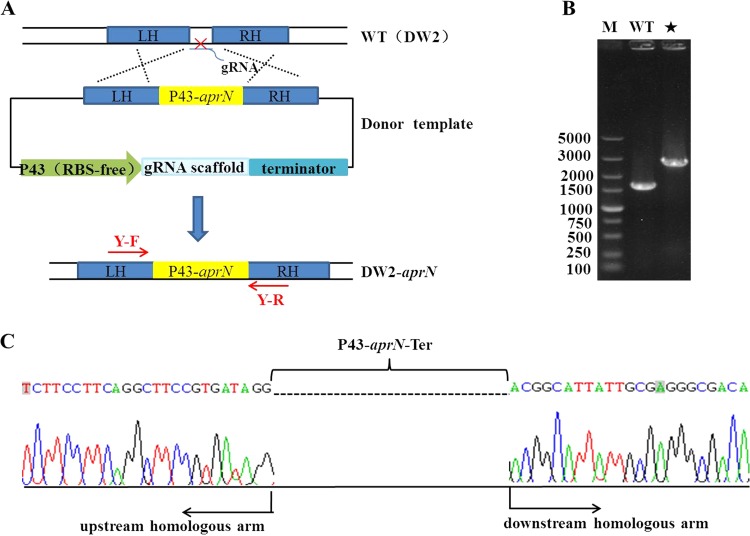
Bacillus strains are important industrial bacteria that can produce various biochemical products. However, low transformation efficiencies and a lack of effective genome editing tools have hindered its widespread application. Recently, clustered regularly interspaced short palindromic repeat (CRISPR)-Cas9 techniques have been utilized in many organisms as genome editing tools because of their high efficiency and easy manipulation. In this study, an efficient genome editing method was developed for Bacillus licheniformis using a CRISPR-Cas9 nickase integrated into the genome of B. licheniformis DW2 with overexpression driven by the P43 promoter. The yvmC gene was deleted using the CRISPR-Cas9n technique with homology arms of 1.0 kb as a representative example, and an efficiency of 100% was achieved. In addition, two genes were simultaneously disrupted with an efficiency of 11.6%, and the large DNA fragment bacABC (42.7 kb) was deleted with an efficiency of 79.0%. Furthermore, the heterologous reporter gene aprN, which codes for nattokinase in Bacillus subtilis, was inserted into the chromosome of B. licheniformis with an efficiency of 76.5%. The activity of nattokinase in the DWc9nΔ7/pP43SNT-SsacC strain reached 59.7 fibrinolytic units (FU)/ml, which was 25.7% higher than that of DWc9n/pP43SNT-SsacC Finally, the engineered strain DWc9nΔ7 (Δepr ΔwprA Δmpr ΔaprE Δvpr ΔbprA ΔbacABC), with multiple disrupted genes, was constructed using the CRISPR-Cas9n technique. Taken together, we have developed an efficient genome editing tool based on CRISPR-Cas9n in B. licheniformis This tool could be applied to strain improvement for future research.IMPORTANCE As important industrial bacteria, Bacillus strains have attracted significant attention due to their production of biological products. However, genetic manipulation of these bacteria is difficult. The CRISPR-Cas9 system has been applied to genome editing in some bacteria, and CRISPR-Cas9n was proven to be an efficient and precise tool in previous reports. The significance of our research is the development of an efficient, more precise, and systematic genome editing method for single-gene deletion, multiple-gene disruption, large DNA fragment deletion, and single-gene integration in Bacillus licheniformis via Cas9 nickase. We also applied this method to the genetic engineering of the host strain for protein expression.
Keywords: Bacillus licheniformis; CRISPR-Cas9n; deletion; genome editing; integration; nattokinase production.
Copyright © 2018 American Society for Microbiology.
Figures






References
-
- Peters JM, Colavin A, Shi H, Czarny TL, Larson MH, Wong S, Hawkins JS, Lu CHS, Koo BM, Marta E, Shiver AL, Whitehead EH, Weissman JS, Brown ED, Qi LS, Huang KC, Gross CA. 2016. A comprehensive, CRISPR-based functional analysis of essential genes in bacteria. Cell 165:1493–1506. doi: 10.1016/j.cell.2016.05.003. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
