

A De Novo FOXP1 Truncating Mutation in a Patient Originally Diagnosed as C Syndrome
- PMID: 29330474
- PMCID: PMC5766530
- DOI: 10.1038/s41598-017-19109-9
A De Novo FOXP1 Truncating Mutation in a Patient Originally Diagnosed as C Syndrome
Abstract
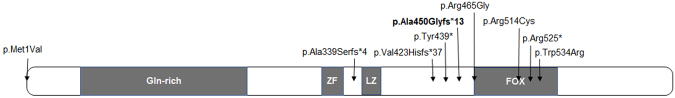
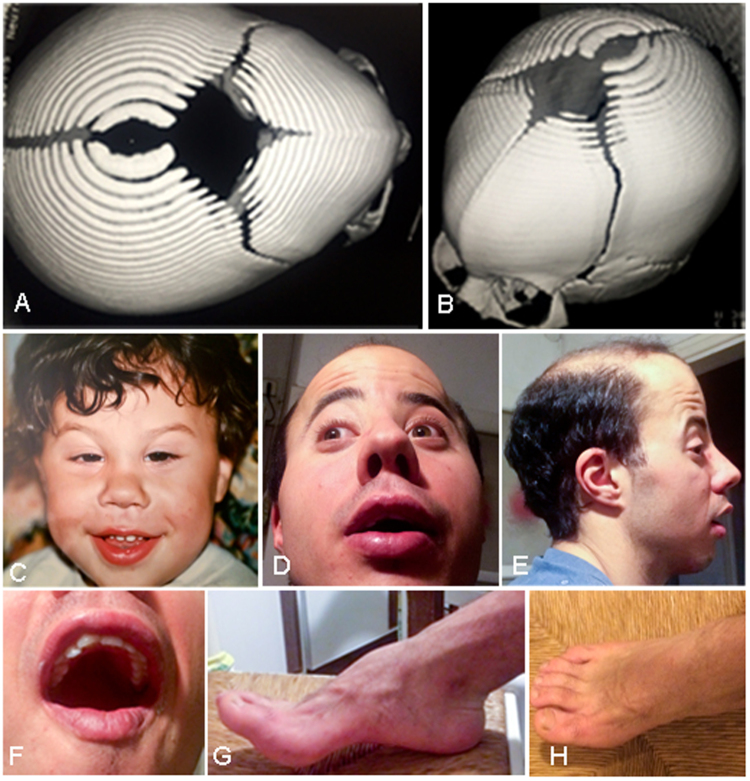
De novo FOXP1 mutations have been associated with intellectual disability (ID), motor delay, autistic features and a wide spectrum of speech difficulties. C syndrome (Opitz C trigonocephaly syndrome) is a rare and genetically heterogeneous condition, characterized by trigonocephaly, craniofacial anomalies and ID. Several different chromosome deletions and and point mutations in distinct genes have been associated with the disease in patients originally diagnosed as Opitz C. By whole exome sequencing we identified a de novo splicing mutation in FOXP1 in a patient, initially diagnosed as C syndrome, who suffers from syndromic intellectual disability with trigonocephaly. The mutation (c.1428 + 1 G > A) promotes the skipping of exon 16, a frameshift and a premature STOP codon (p.Ala450GLyfs*13), as assessed by a minigene strategy. The patient reported here shares speech difficulties, intellectual disability and autistic features with other FOXP1 syndrome patients, and thus the diagnosis for this patient should be changed. Finally, since trigonocephaly has not been previously reported in FOXP1 syndrome, it remains to be proved whether it may be associated with the FOXP1 mutation.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Tetreault M, Bareke E, Nadaf J, Alirezaie N, Majewski J. Whole-exome sequencing as a diagnostic tool: current challenges and future opportunities. Expert Rev Mol Diagn. 2015;15:749–760. - PubMed
