

Classification and evolution of human papillomavirus genome variants: Alpha-5 (HPV26, 51, 69, 82), Alpha-6 (HPV30, 53, 56, 66), Alpha-11 (HPV34, 73), Alpha-13 (HPV54) and Alpha-3 (HPV61)
- PMID: 29331867
- PMCID: PMC6093212
- DOI: 10.1016/j.virol.2018.01.002
Classification and evolution of human papillomavirus genome variants: Alpha-5 (HPV26, 51, 69, 82), Alpha-6 (HPV30, 53, 56, 66), Alpha-11 (HPV34, 73), Alpha-13 (HPV54) and Alpha-3 (HPV61)
Abstract
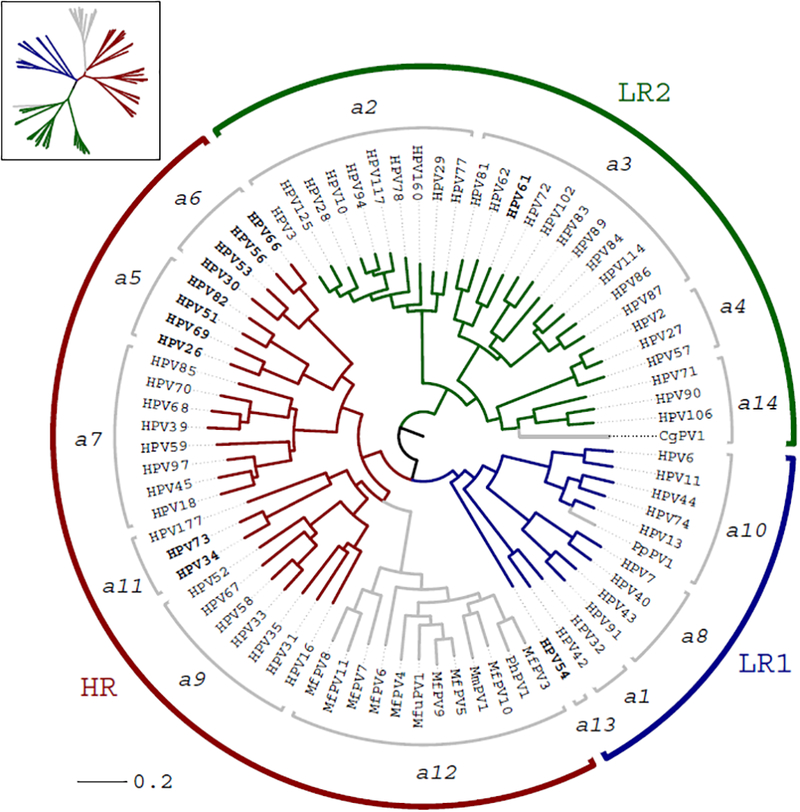
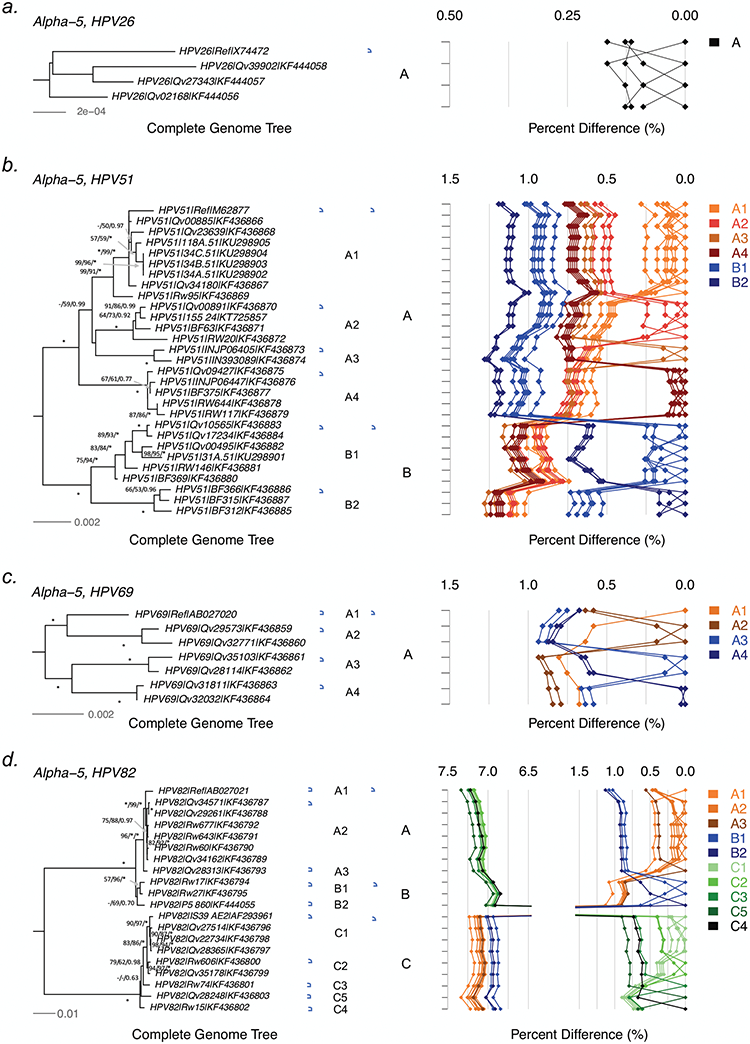
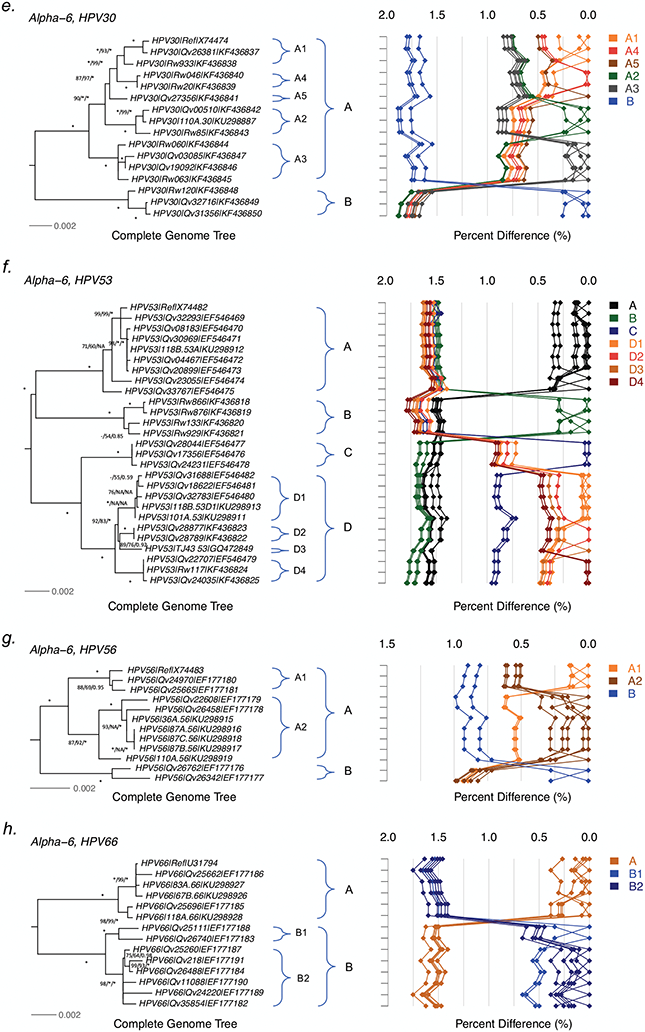
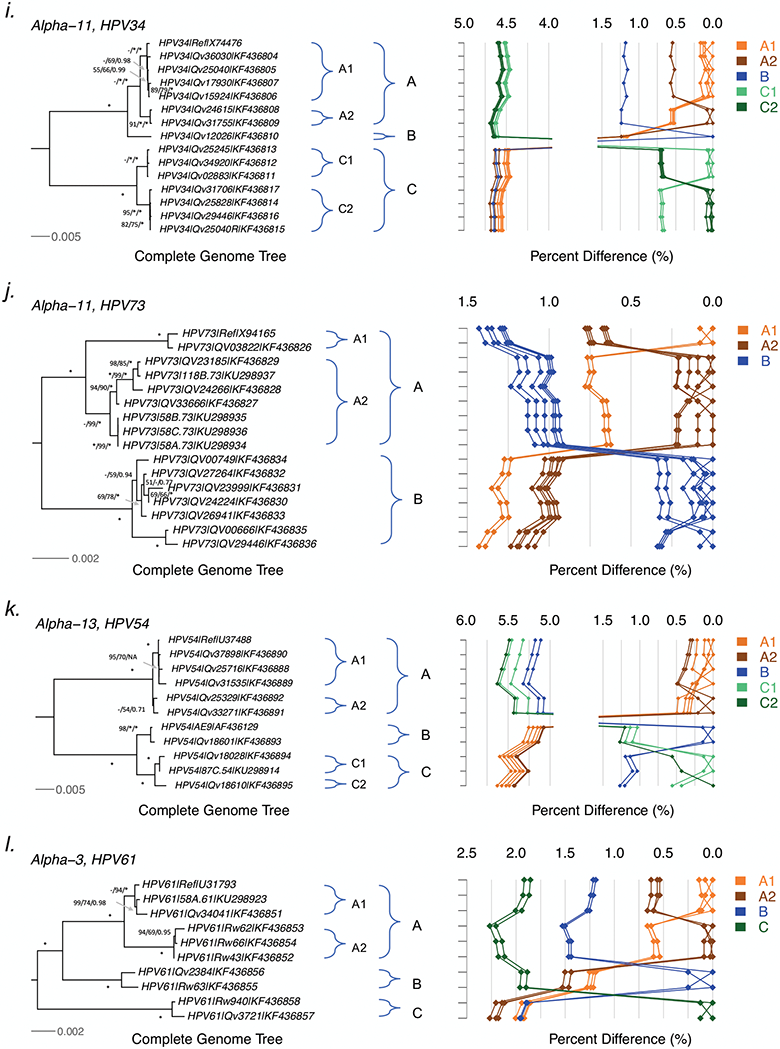
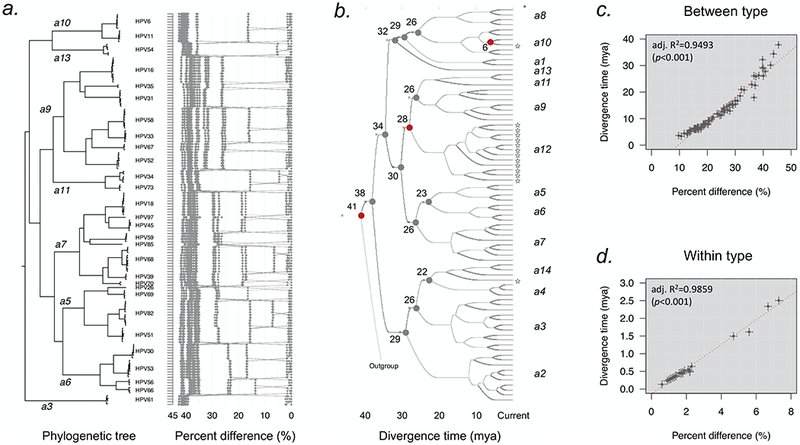
HPV variants from the same type can be classified into lineages and sublineages based on the complete genome differences and the phylogenetic topologies. We examined nucleotide variations of twelve HPV types within the species Alpha-5 (HPV26, 51, 69, 82), Alpha-6 (HPV30, 53, 56, 66), Alpha-11 (HPV34, 73), Alpha-13 (HPV54) and Alpha-3 (HPV61) by analyzing 1432 partial sequences and 181 complete genomes from multiple geographic populations. The inter-lineage and inter-sublineage mean differences of HPV variants ranged between 0.9-7.3% and 0.3-0.9%, respectively. The heterogeneity and phylogenies of HPV isolates indicate an independent evolutionary history for each type. The noncoding regions were the most variable regions whereas the capsid proteins were relatively conserved. Certain variant lineages and/or sublineages were geographically-associated. These data provide the basis to further classify HPV variants and should foster future studies on the evolution of HPV genomes and the associations of HPV variants with cancer risk.
Keywords: Cervical cancer; Classification; Evolution; Human papillomavirus; Variant.
Copyright © 2018 Elsevier Inc. All rights reserved.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
