

Charge migration and charge transfer in molecular systems
- PMID: 29333473
- PMCID: PMC5745195
- DOI: 10.1063/1.4996505
Charge migration and charge transfer in molecular systems
Abstract
The transfer of charge at the molecular level plays a fundamental role in many areas of chemistry, physics, biology and materials science. Today, more than 60 years after the seminal work of R. A. Marcus, charge transfer is still a very active field of research. An important recent impetus comes from the ability to resolve ever faster temporal events, down to the attosecond time scale. Such a high temporal resolution now offers the possibility to unravel the most elementary quantum dynamics of both electrons and nuclei that participate in the complex process of charge transfer. This review covers recent research that addresses the following questions. Can we reconstruct the migration of charge across a molecule on the atomic length and electronic time scales? Can we use strong laser fields to control charge migration? Can we temporally resolve and understand intramolecular charge transfer in dissociative ionization of small molecules, in transition-metal complexes and in conjugated polymers? Can we tailor molecular systems towards specific charge-transfer processes? What are the time scales of the elementary steps of charge transfer in liquids and nanoparticles? Important new insights into each of these topics, obtained from state-of-the-art ultrafast spectroscopy and/or theoretical methods, are summarized in this review.
Figures
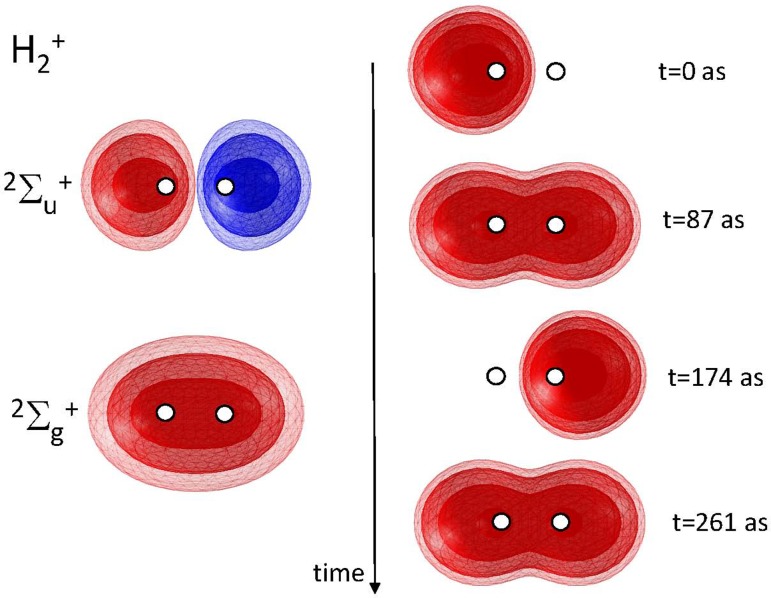
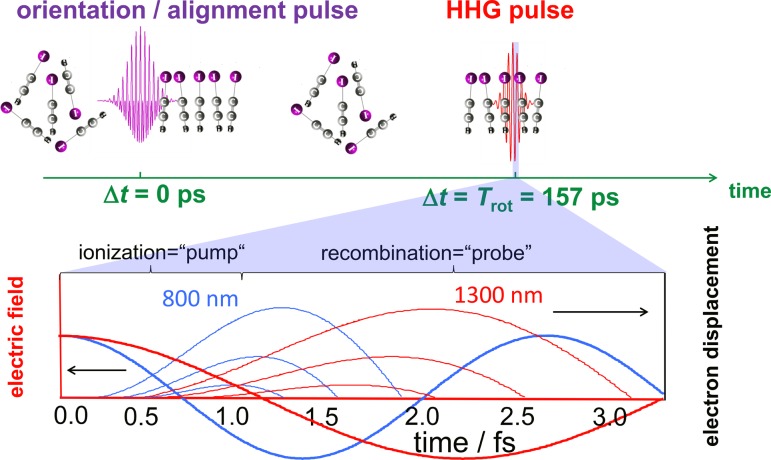
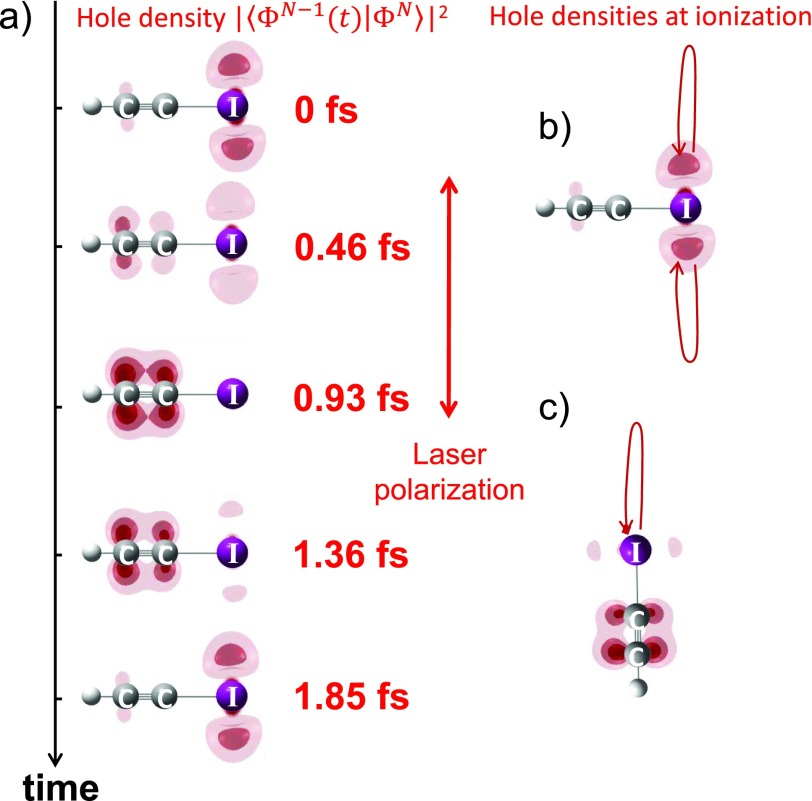
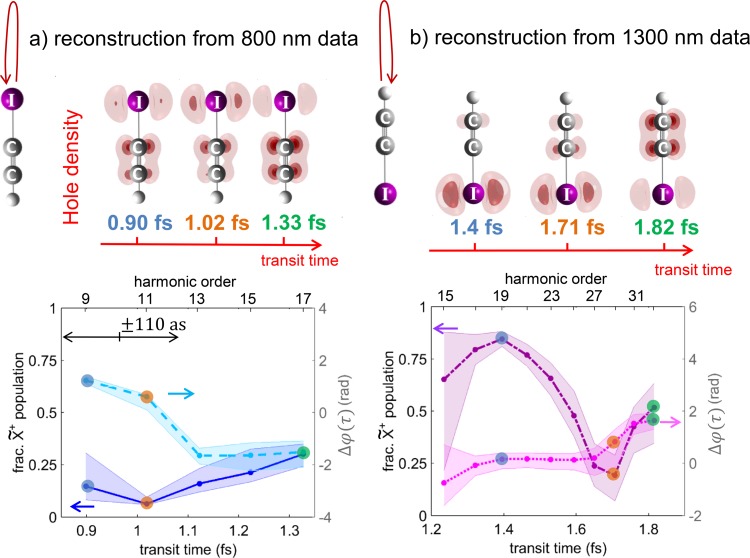
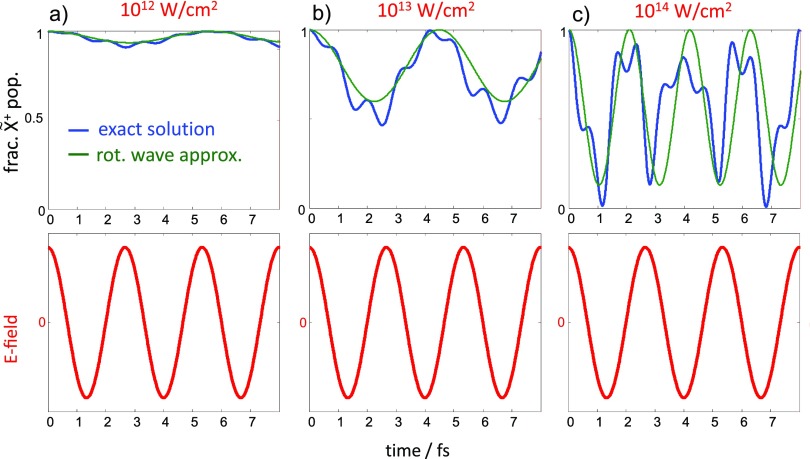
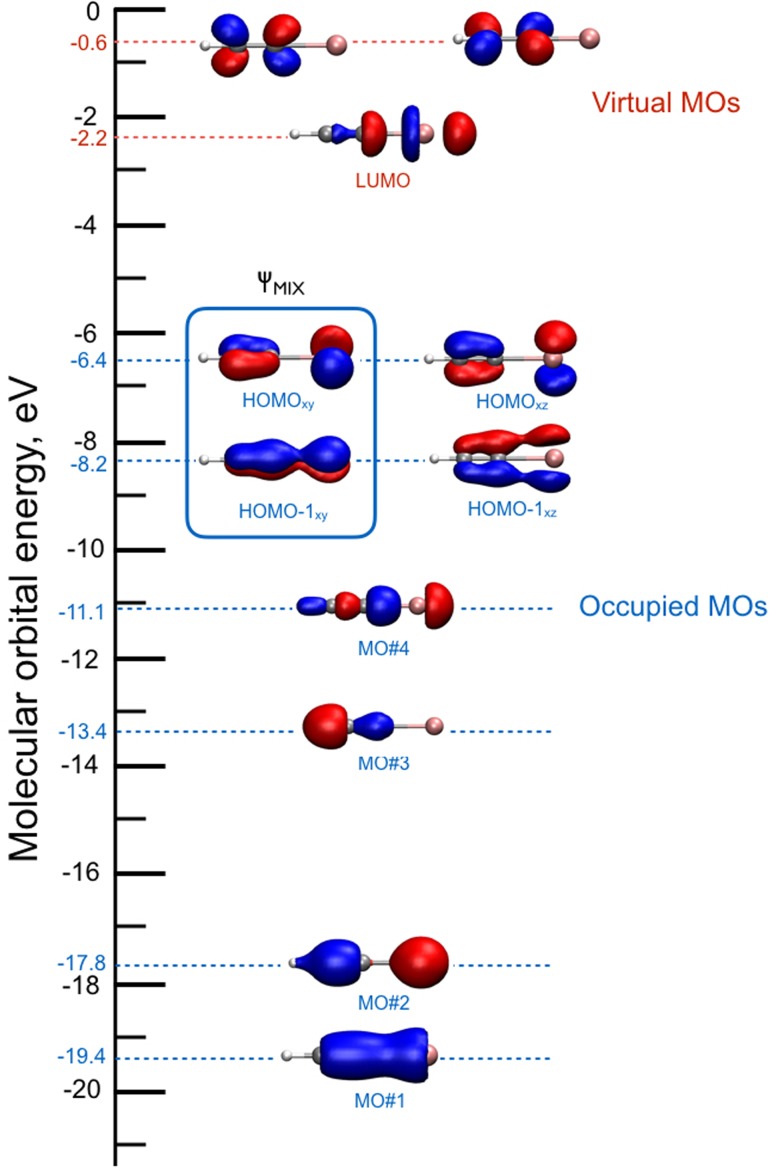
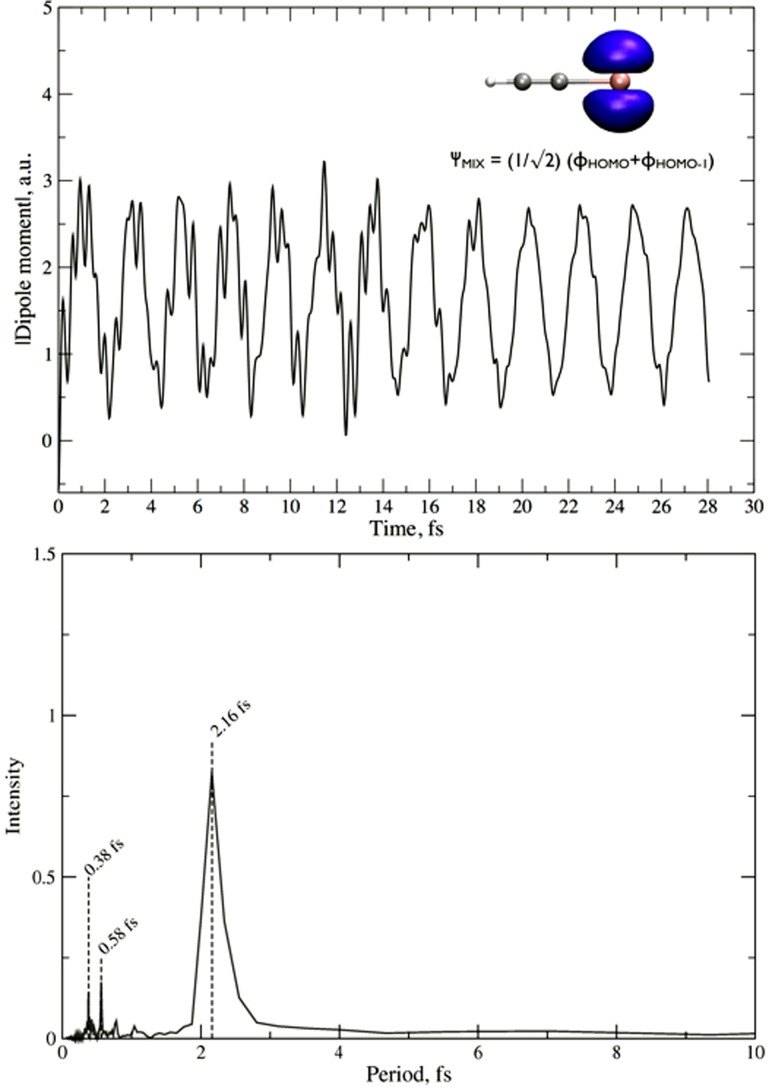
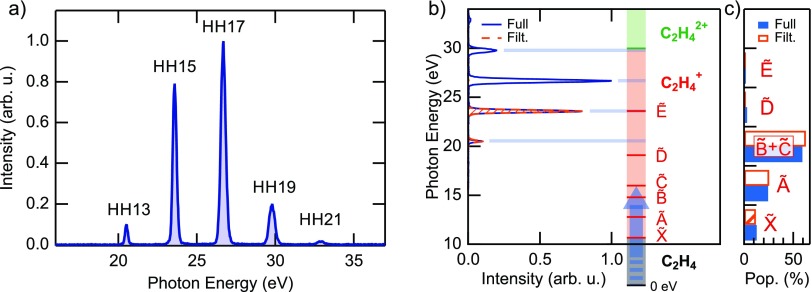
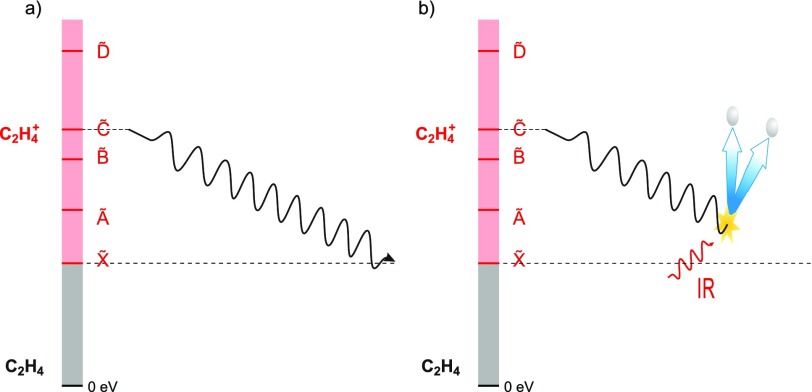
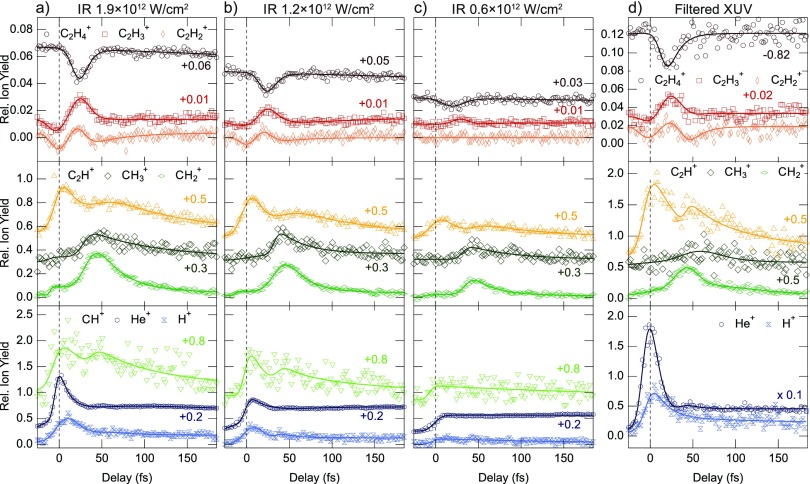
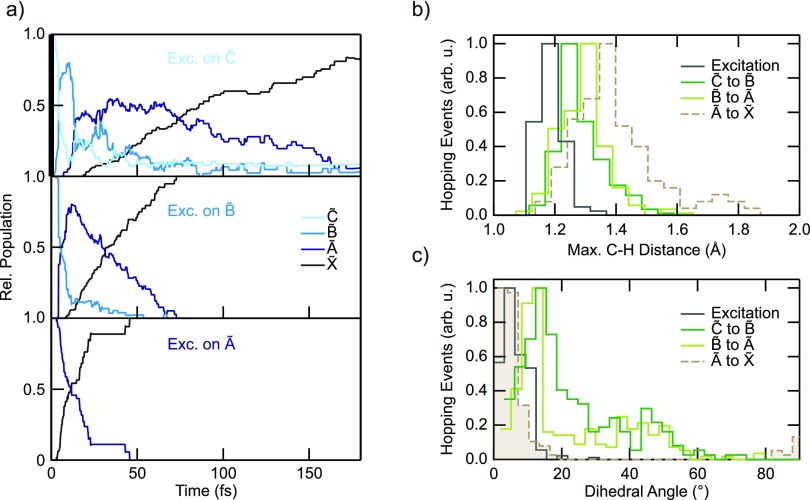
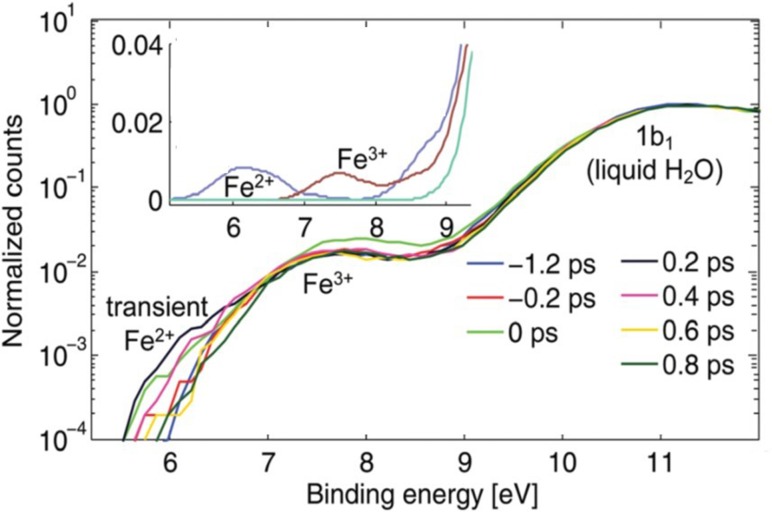
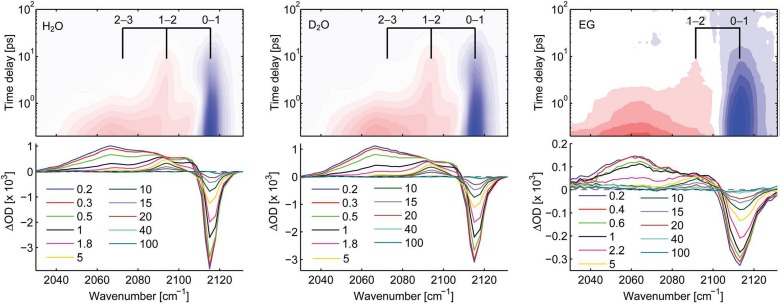
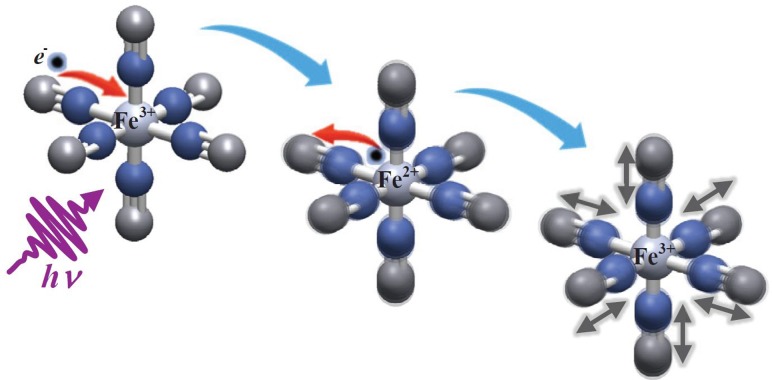
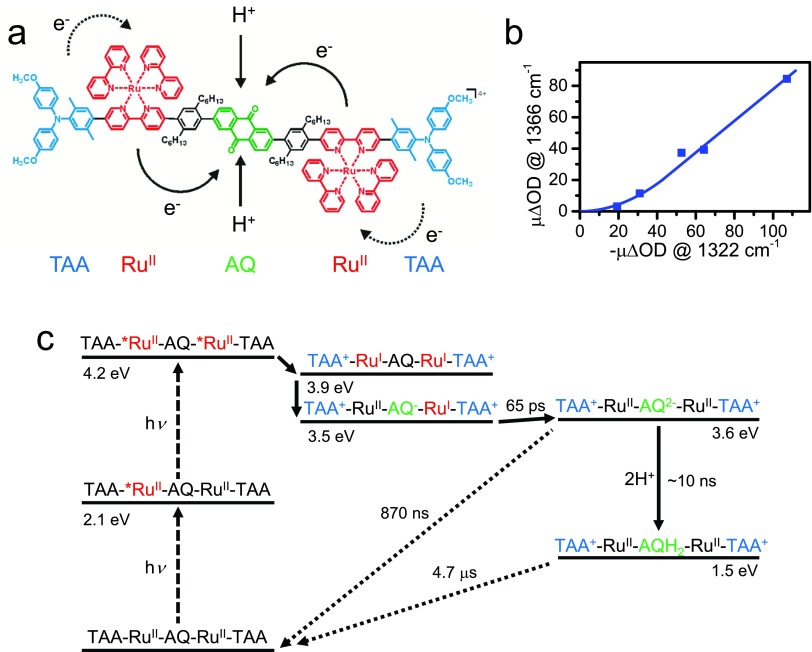
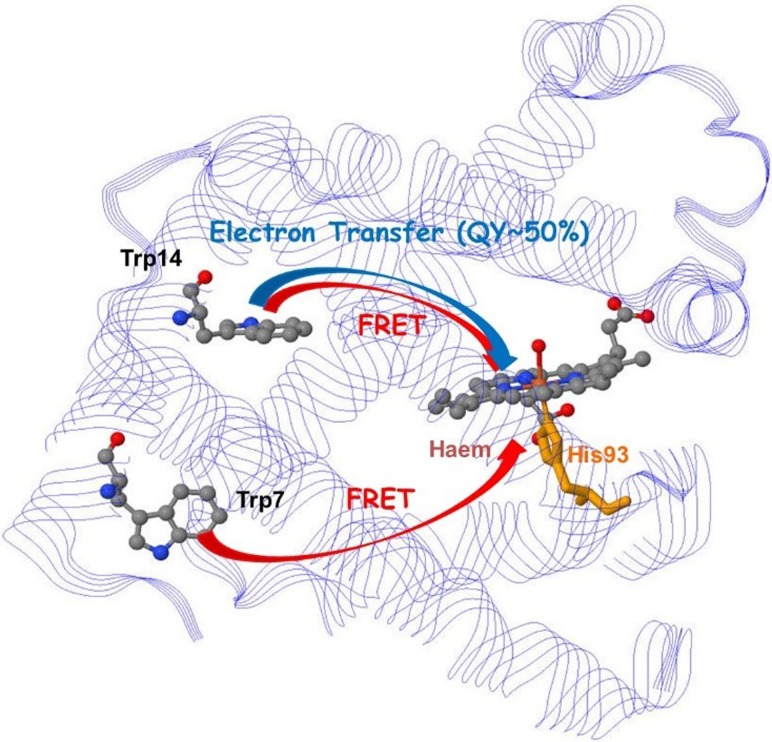
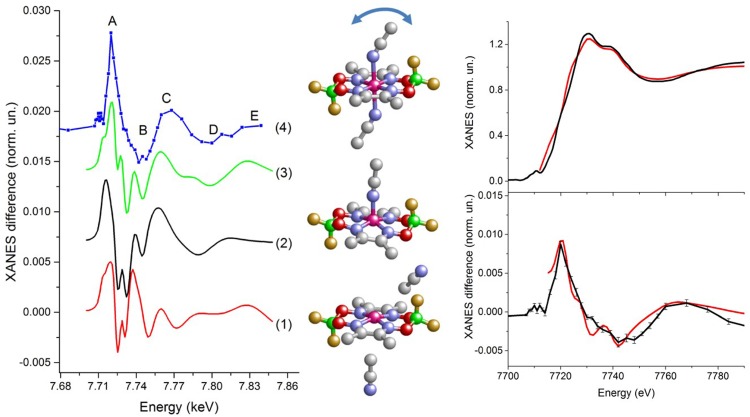
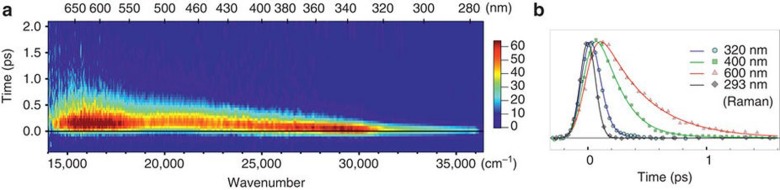
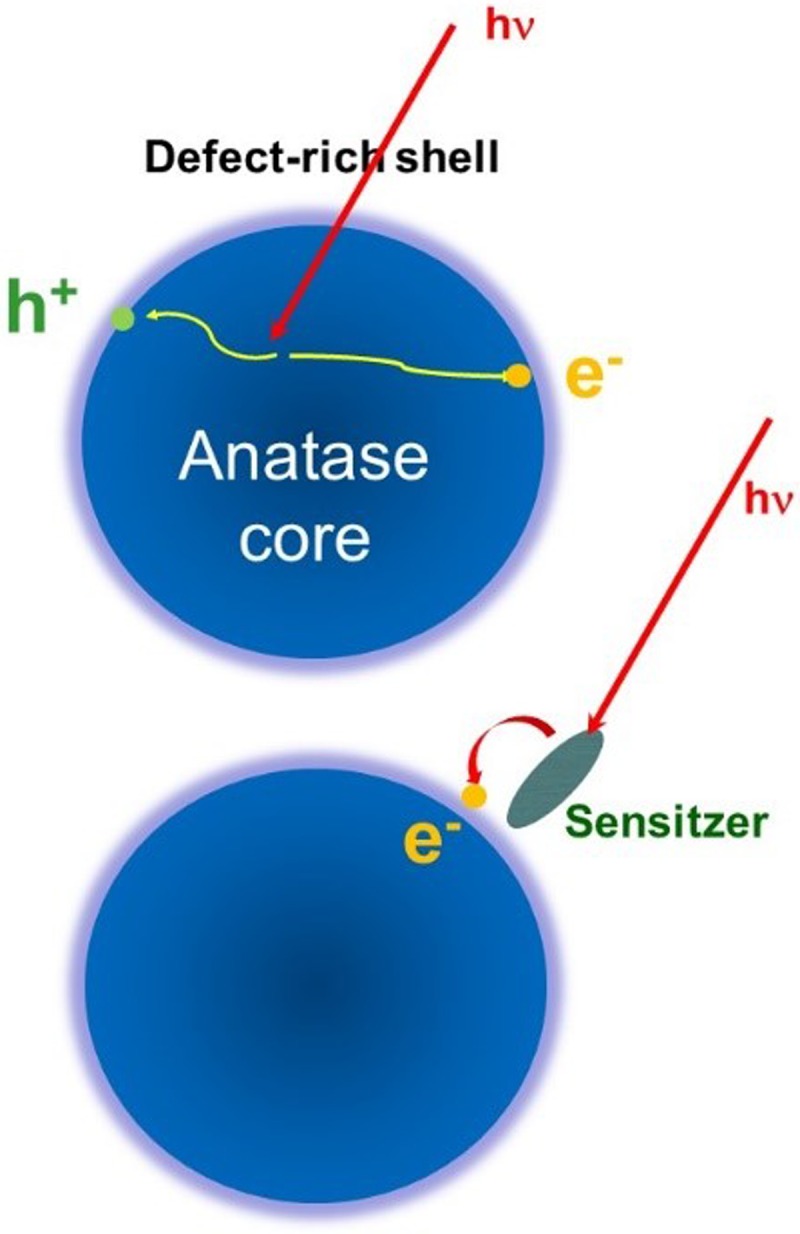
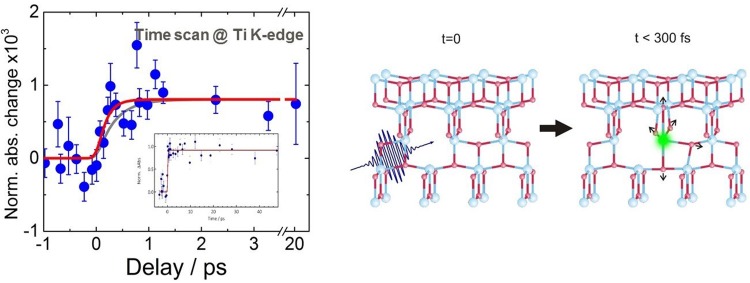
References
-
- May V. and Kühn O., Charge and Energy Transfer Dynamics in Molecular Systems ( John Wiley & Sons, 2011).
-
- Marcus R. A., J. Chem. Phys. 24, 966 (1956). 10.1063/1.1742723 - DOI
-
- Marcus R. A., J. Chem. Phys. 24, 979 (1956). 10.1063/1.1742724 - DOI
-
- Zewail A. H., J. Phys. Chem. A 104, 5660 (2000). 10.1021/jp001460h - DOI
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources