

In-depth resistome analysis by targeted metagenomics
- PMID: 29335005
- PMCID: PMC5769438
- DOI: 10.1186/s40168-017-0387-y
In-depth resistome analysis by targeted metagenomics
Abstract
Background: Antimicrobial resistance is a major global health challenge. Metagenomics allows analyzing the presence and dynamics of "resistomes" (the ensemble of genes encoding antimicrobial resistance in a given microbiome) in disparate microbial ecosystems. However, the low sensitivity and specificity of available metagenomic methods preclude the detection of minority populations (often present below their detection threshold) and/or the identification of allelic variants that differ in the resulting phenotype. Here, we describe a novel strategy that combines targeted metagenomics using last generation in-solution capture platforms, with novel bioinformatics tools to establish a standardized framework that allows both quantitative and qualitative analyses of resistomes.
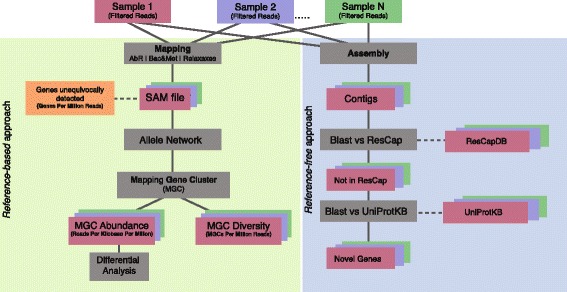
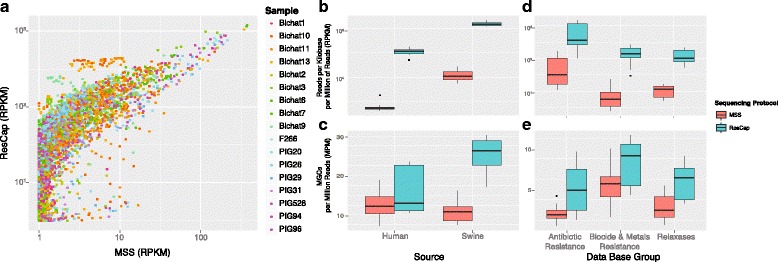
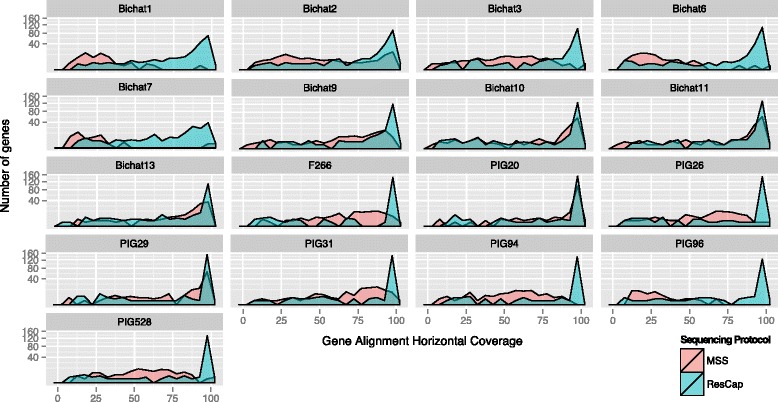
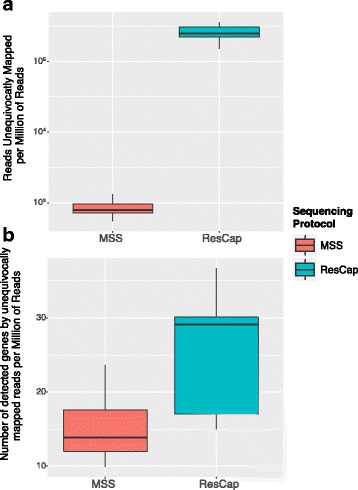
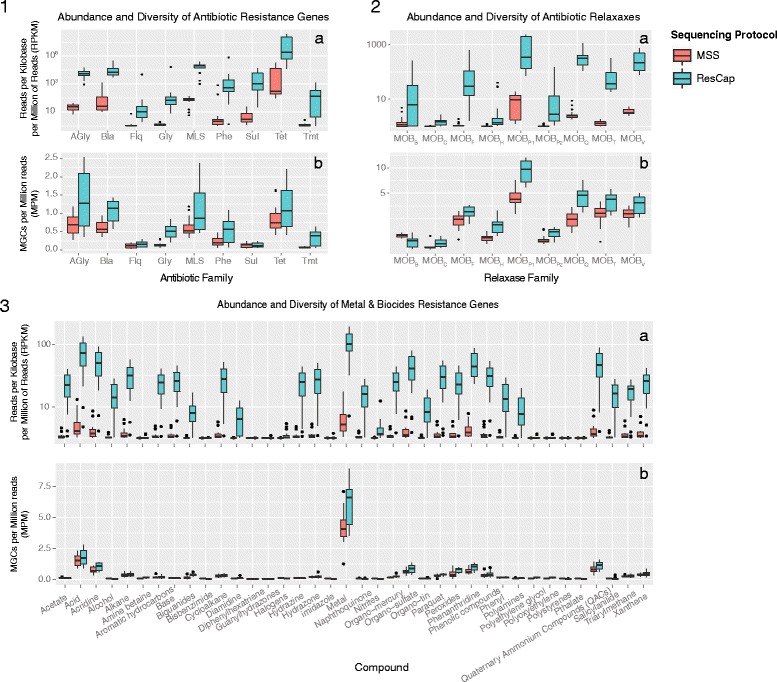
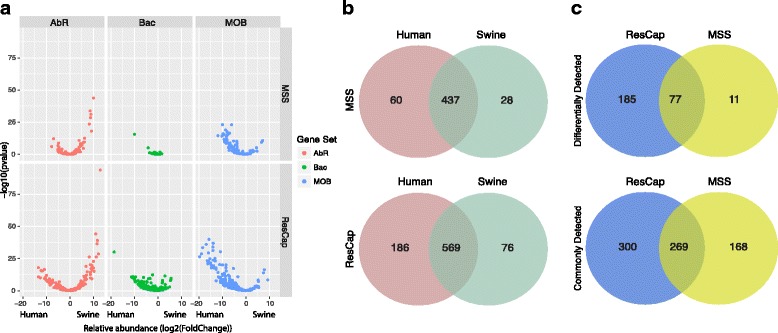
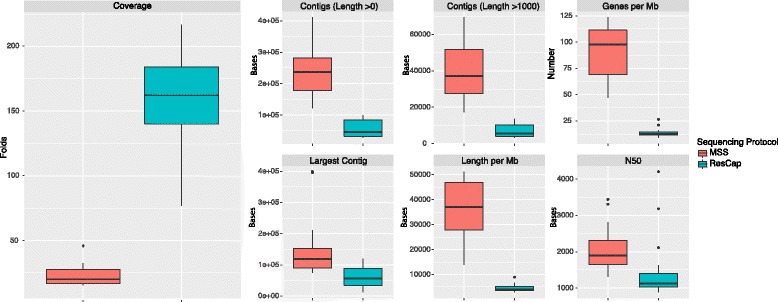
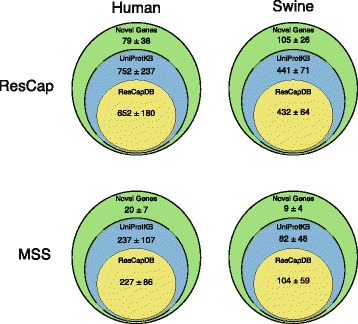
Methods: We developed ResCap, a targeted sequence capture platform based on SeqCapEZ (NimbleGene) technology, which includes probes for 8667 canonical resistance genes (7963 antibiotic resistance genes and 704 genes conferring resistance to metals or biocides), and 2517 relaxase genes (plasmid markers) and 78,600 genes homologous to the previous identified targets (47,806 for antibiotics and 30,794 for biocides or metals). Its performance was compared with metagenomic shotgun sequencing (MSS) for 17 fecal samples (9 humans, 8 swine). ResCap significantly improves MSS to detect "gene abundance" (from 2.0 to 83.2%) and "gene diversity" (26 versus 14.9 genes unequivocally detected per sample per million of reads; the number of reads unequivocally mapped increasing up to 300-fold by using ResCap), which were calculated using novel bioinformatic tools. ResCap also facilitated the analysis of novel genes potentially involved in the resistance to antibiotics, metals, biocides, or any combination thereof.
Conclusions: ResCap, the first targeted sequence capture, specifically developed to analyze resistomes, greatly enhances the sensitivity and specificity of available metagenomic methods and offers the possibility to analyze genes related to the selection and transfer of antimicrobial resistance (biocides, heavy metals, plasmids). The model opens the possibility to study other complex microbial systems in which minority populations play a relevant role.
Keywords: Antimicrobial resistance; Differential abundance analysis; Metagenomics; Resistome; Targeted metagenomics.
Conflict of interest statement
Ethics approval and consent to participate
Clinical samples were used after obtaining the approval of the study by the Institutional Review Board of the Hospital Bichat, and all written informed consents from the enrolled subjects, in compliance with national legislation and the Code of Ethical Principles for Medical Research Involving Human Subjects of the World Medical Association (Declaration of Helsinki).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures

References
-
- Årdal C, Outterson K, Hoffman SJ, Ghafur A, Sharland M, Ranganathan N, Smith R, Zorzet A, Cohn J, Pittet D, Daulaire N, Morel C, Rizvi Z, Balasegaram M, Dar OA, Heymann DL, Holmes AH, Moore LS, Laxminarayan R, Mendelson M, Røttingen JA. International cooperation to improve access to and sustain effectiveness of antimicrobials. Lancet. 2016;387:296–307. doi: 10.1016/S0140-6736(15)00470-5. - DOI - PubMed
-
- Årdal C, Baraldi E, Ciabuschi F, Outterson K, Rex JH, Piddock LJV, Findlay D; DRIVE-AB Steering Committee. To the G20: incentivising antibacterial research and development. Lancet Infect Dis. 2017:799-801. - PubMed
-
- Dar OA, Hasan R, Schlundt J, Harbarth S, Caleo G, Dar FK, Littmann J, Rweyemamu M, Buckley EJ, Shahid M, Kock R, Li HL, Giha H, Khan M, So AD, Bindayna KM, Kessel A, Pedersen HB, Permanand G, Zumla A, Røttingen JA, Heymann DL. Exploring the evidence base for national and regional policy interventions to combat resistance. Lancet. 2016;387(10015):285-95. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- PLASWIRES-612146/FP7-ICT- 2013-10/Ministry of Economy and Competitiveness of Spain/International
- PI15-00818/Spanish R&D National Plan 2012-2019/International
- PROMPT-S2010/BMD2414/Regional Government of Madrid/International
- BIO2014-54507-R/Ministry of Economy and Competitiveness of Spa/International
- L60 MD002414/MD/NIMHD NIH HHS/United States
- RD12/0015/Spanish Network for Research on Infectious Diseases (REIPI)/International
- BFU2014-55534- C2-1-P/Spanish R&D National Plan 2012-2019/International
- BFU2014-55534-C2-1-P/Ministry of Economy and Competitiveness of Spain/International
- ANR-11-DPBS-0001/Metagenopolis grant/International
- BIO2014-54507-R/Spanish R&D National Plan 2012-2019/International
- JPIW2013-089-C02-01/JPI WATER STARE/International
- CB06/02/0053/CIBER in Epidemiology and Public Health, CIBERESP/International
- PI15-0512/Spanish R&D National Plan 2012-2019/International
- 282004/EVOTARFP7-HEALTH/International
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
