

PD-L1 on host cells is essential for PD-L1 blockade-mediated tumor regression
- PMID: 29337303
- PMCID: PMC5785245
- DOI: 10.1172/JCI96061
PD-L1 on host cells is essential for PD-L1 blockade-mediated tumor regression
Abstract
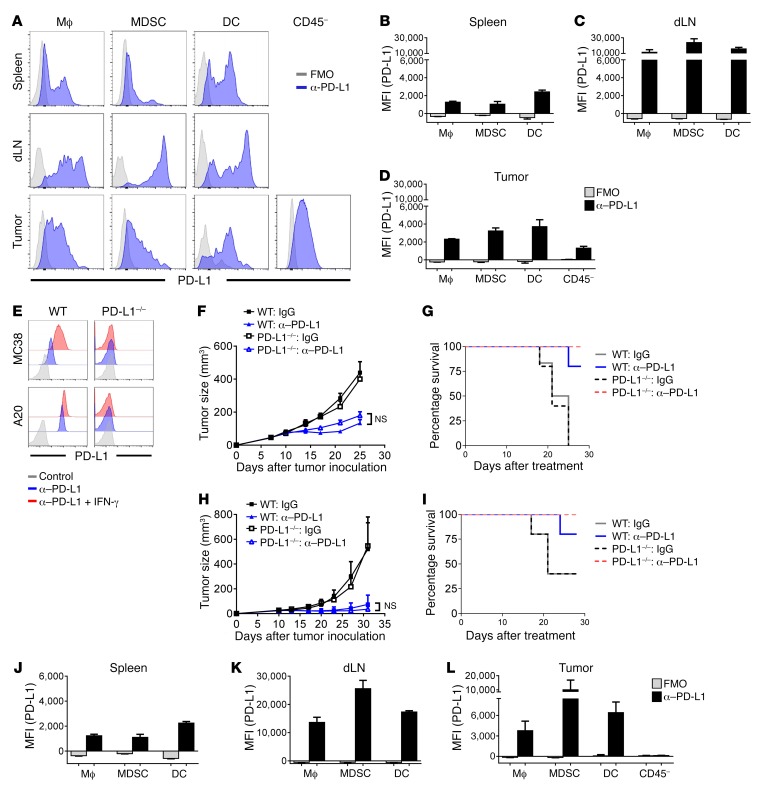
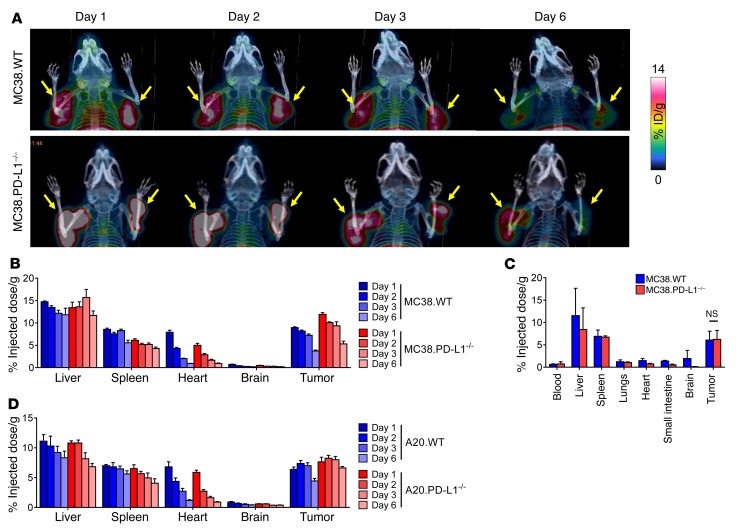
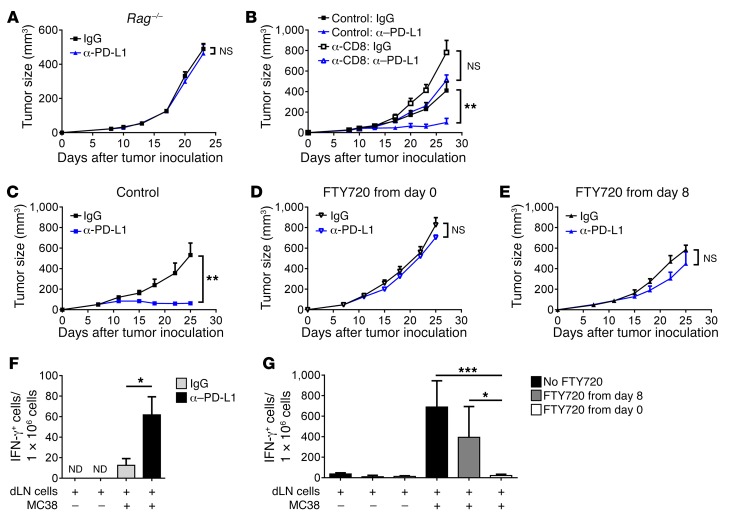
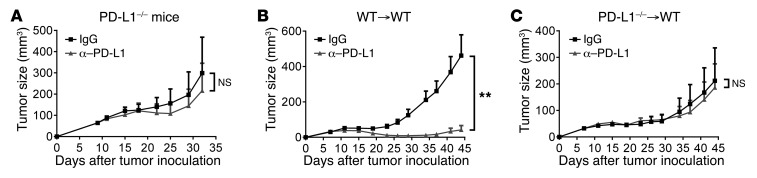
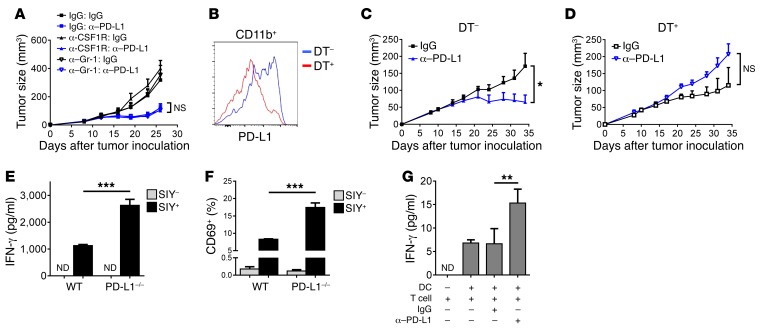
Programmed death-ligand 1 (PD-L1) expression on tumor cells is essential for T cell impairment, and PD-L1 blockade therapy has shown unprecedented durable responses in several clinical studies. Although higher expression of PD-L1 on tumor cells is associated with a better immune response after Ab blockade, some PD-L1-negative patients also respond to this therapy. In the current study, we explored whether PD-L1 on tumor or host cells was essential for anti-PD-L1-mediated therapy in 2 different murine tumor models. Using real-time imaging in whole tumor tissues, we found that anti-PD-L1 Ab accumulates in tumor tissues, regardless of the status of PD-L1 expression on tumor cells. We further observed that, while PD-L1 on tumor cells was largely dispensable for the response to checkpoint blockade, PD-L1 in host myeloid cells was essential for this response. Additionally, PD-L1 signaling in defined antigen-presenting cells (APCs) negatively regulated and inhibited T cell activation. PD-L1 blockade inside tumors was not sufficient to mediate regression, as limiting T cell trafficking reduced the efficacy of the blockade. Together, these findings demonstrate that PD-L1 expressed in APCs, rather than on tumor cells, plays an essential role in checkpoint blockade therapy, providing an insight into the mechanisms of this therapy.
Keywords: Cancer immunotherapy; Cellular immune response; Immunology; Oncology.
Conflict of interest statement
Figures
Comment in
-
The host protecting the tumor from the host - targeting PD‑L1 expressed by host cells.J Clin Invest. 2018 Feb 1;128(2):570-572. doi: 10.1172/JCI99047. Epub 2018 Jan 16. J Clin Invest. 2018. PMID: 29337304 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
