

Subcellular Reactive Oxygen Species (ROS) in Cardiovascular Pathophysiology
- PMID: 29337890
- PMCID: PMC5789324
- DOI: 10.3390/antiox7010014
Subcellular Reactive Oxygen Species (ROS) in Cardiovascular Pathophysiology
Abstract
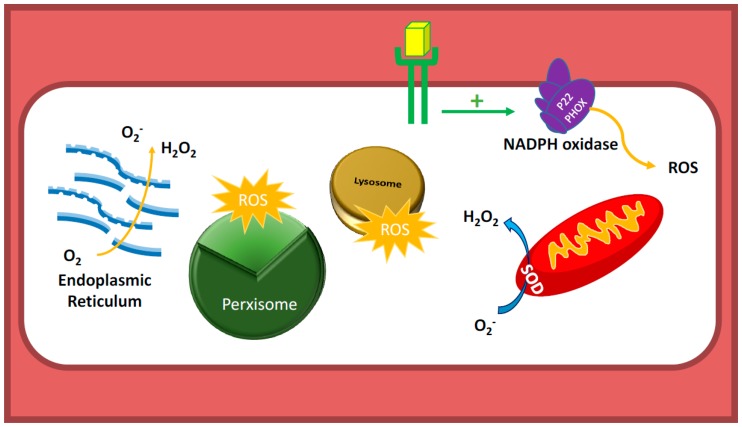
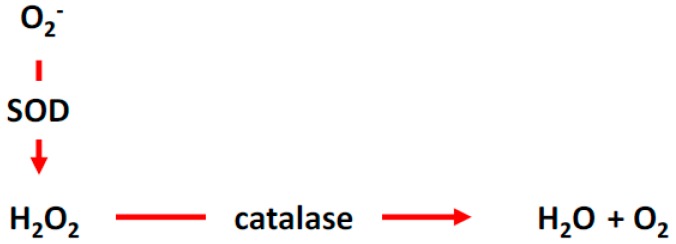
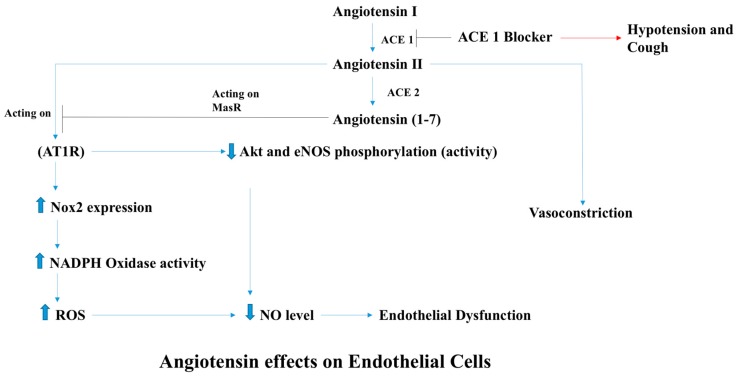
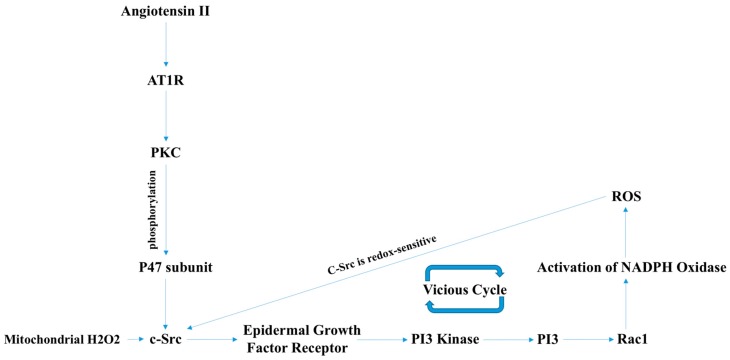
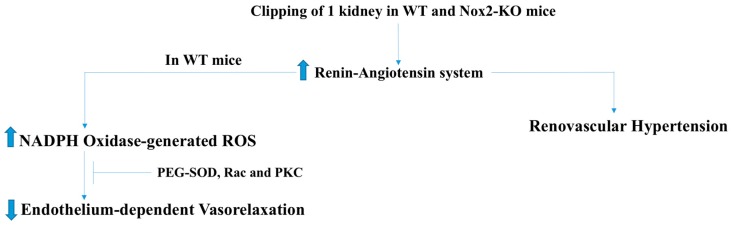
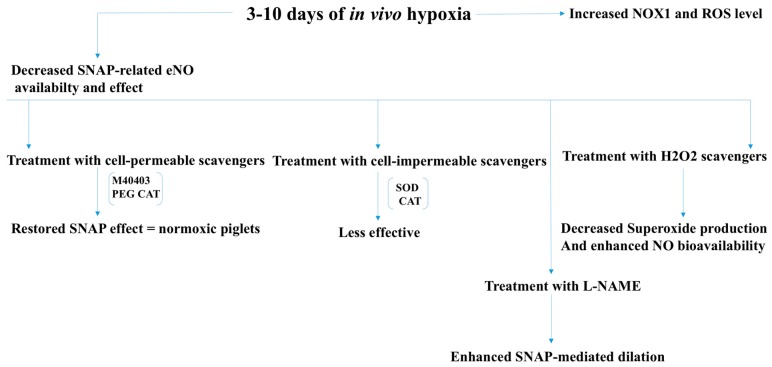
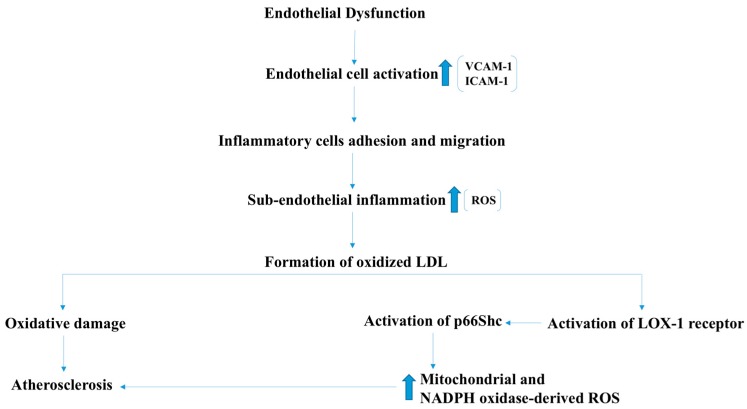
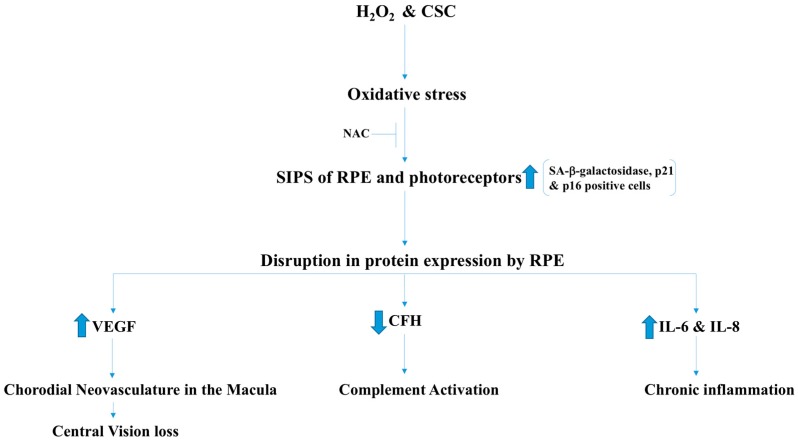
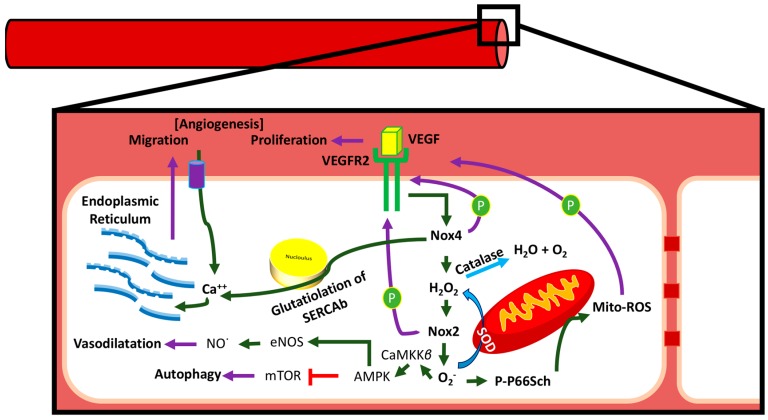
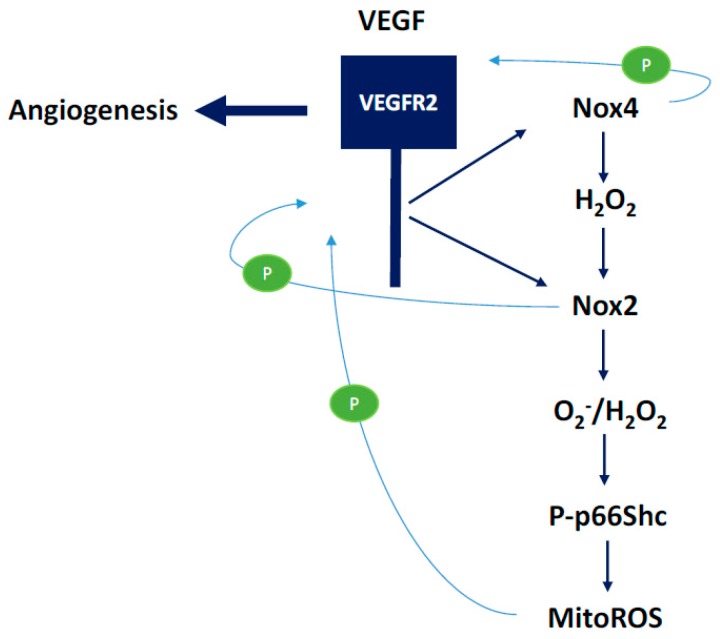
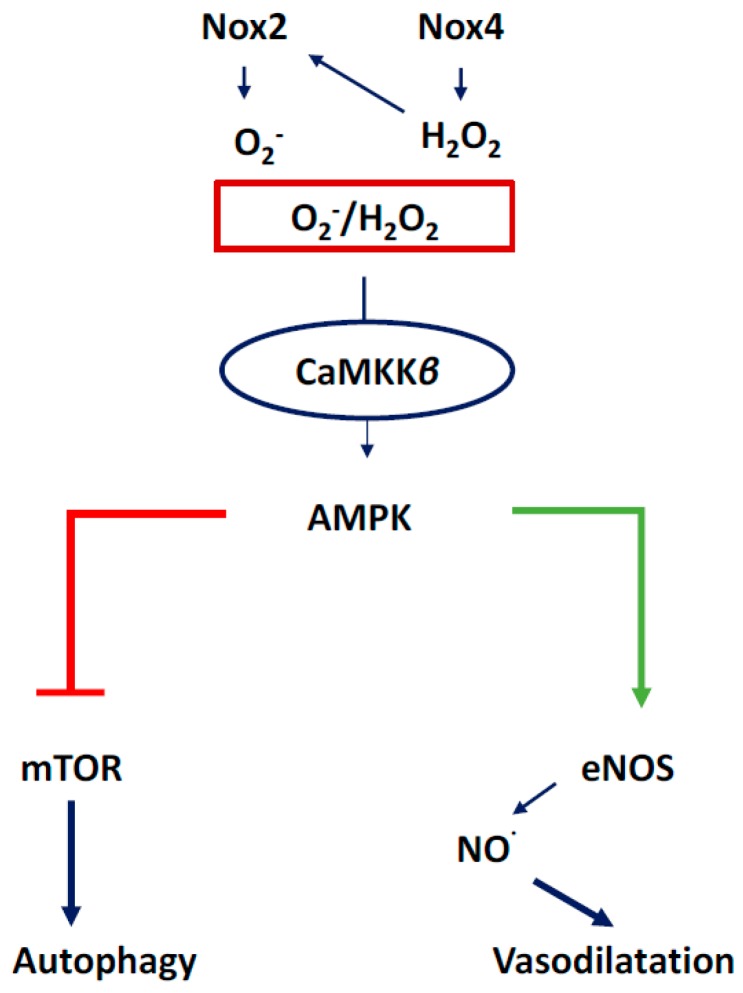
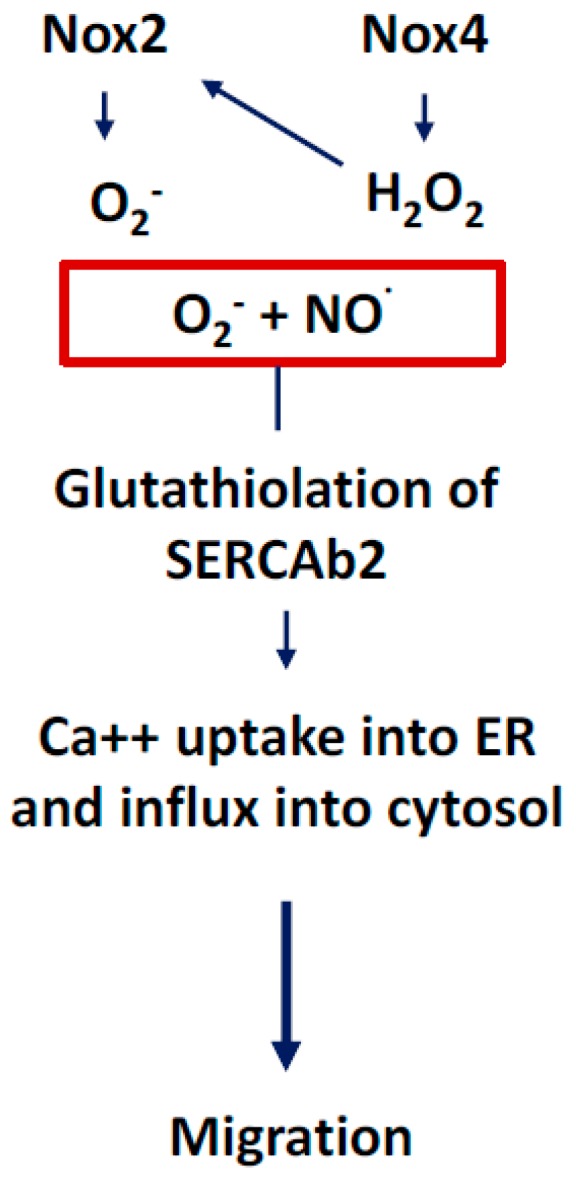
There exist two opposing perspectives regarding reactive oxygen species (ROS) and their roles in angiogenesis and cardiovascular system, one that favors harmful and causal effects of ROS, while the other supports beneficial effects. Recent studies have shown that interaction between ROS in different sub-cellular compartments plays a crucial role in determining the outcomes (beneficial vs. deleterious) of ROS exposures on the vascular system. Oxidant radicals in one cellular organelle can affect the ROS content and function in other sub-cellular compartments in endothelial cells (ECs). In this review, we will focus on a critical fact that the effects or the final phenotypic outcome of ROS exposure to EC are tissue- or organ-specific, and depend on the spatial (subcellular localization) and temporal (duration of ROS exposure) modulation of ROS levels.
Keywords: Nicotinamide Adenine Dinucleotide Phosphate (NADPH) oxidase; ROS; angiogenesis; cardiovascular disease; coronary endothelium; mitochondrial ROS.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
