

The impact of ribosomal interference, codon usage, and exit tunnel interactions on translation elongation rate variation
- PMID: 29337993
- PMCID: PMC5786338
- DOI: 10.1371/journal.pgen.1007166
The impact of ribosomal interference, codon usage, and exit tunnel interactions on translation elongation rate variation
Erratum in
-
Correction: The impact of ribosomal interference, codon usage, and exit tunnel interactions on translation elongation rate variation.PLoS Genet. 2018 Aug 24;14(8):e1007620. doi: 10.1371/journal.pgen.1007620. eCollection 2018 Aug. PLoS Genet. 2018. PMID: 30142215 Free PMC article.
Abstract
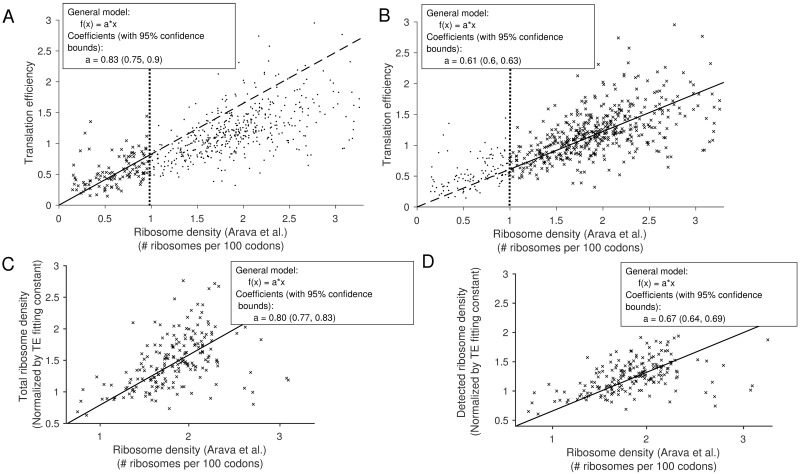
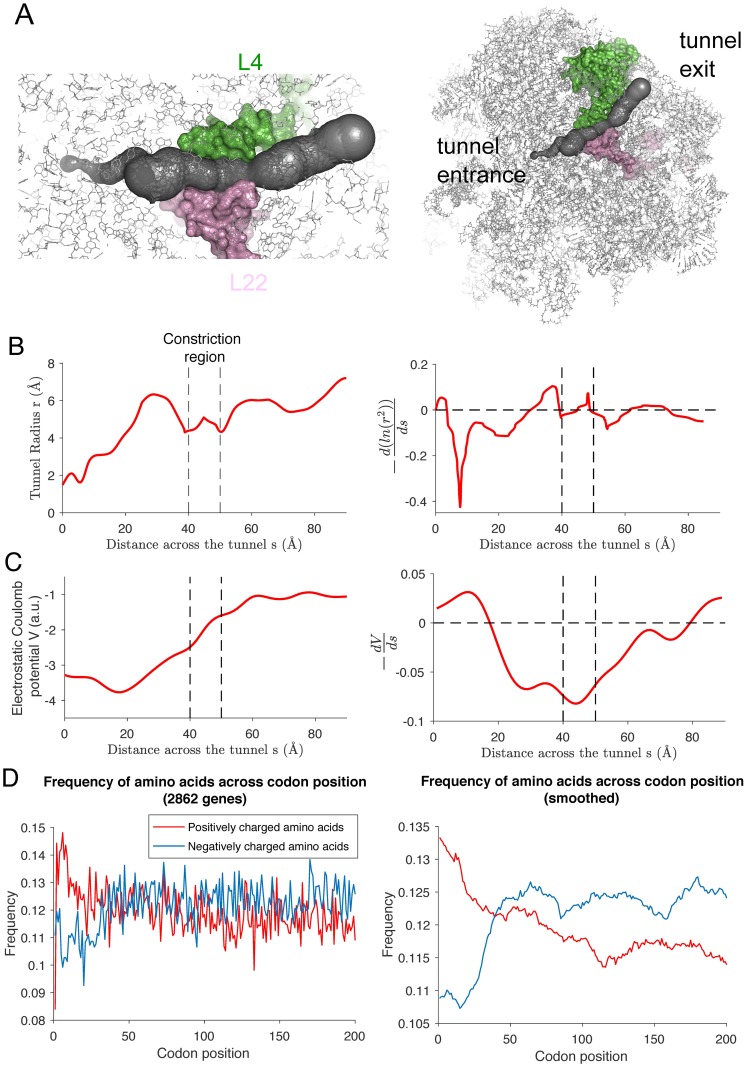
Previous studies have shown that translation elongation is regulated by multiple factors, but the observed heterogeneity remains only partially explained. To dissect quantitatively the different determinants of elongation speed, we use probabilistic modeling to estimate initiation and local elongation rates from ribosome profiling data. This model-based approach allows us to quantify the extent of interference between ribosomes on the same transcript. We show that neither interference nor the distribution of slow codons is sufficient to explain the observed heterogeneity. Instead, we find that electrostatic interactions between the ribosomal exit tunnel and specific parts of the nascent polypeptide govern the elongation rate variation as the polypeptide makes its initial pass through the tunnel. Once the N-terminus has escaped the tunnel, the hydropathy of the nascent polypeptide within the ribosome plays a major role in modulating the speed. We show that our results are consistent with the biophysical properties of the tunnel.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures





References
-
- Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324(5924):218–223. doi: 10.1126/science.1168978 - DOI - PMC - PubMed
-
- Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nature Protocols. 2012;7(8):1534–1550. doi: 10.1038/nprot.2012.086 - DOI - PMC - PubMed
-
- Ingolia NT. Ribosome profiling: new views of translation, from single codons to genome scale. Nature Reviews Genetics. 2014;15(3):205–213. doi: 10.1038/nrg3645 - DOI - PubMed
-
- Vogel C, Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nature Reviews Genetics. 2012;13(4):227–232. doi: 10.1038/nrg3185 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
