

Kinase-independent function of E-type cyclins in liver cancer
- PMID: 29339491
- PMCID: PMC5798328
- DOI: 10.1073/pnas.1711477115
Kinase-independent function of E-type cyclins in liver cancer
Abstract
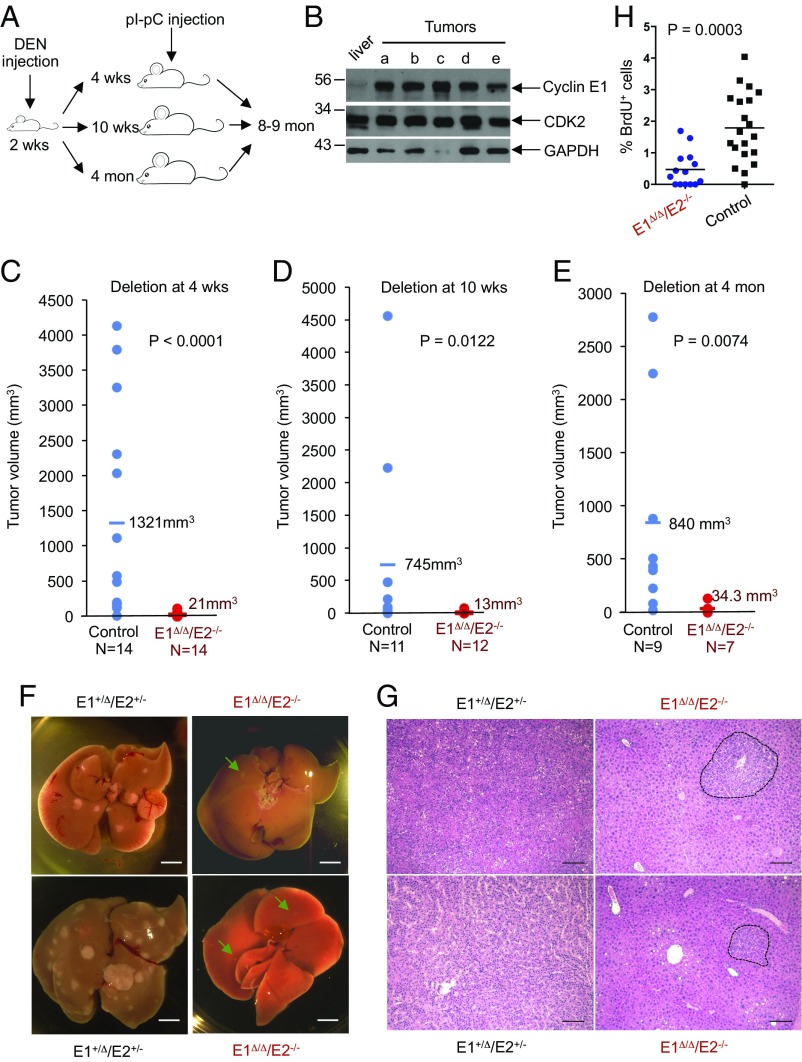
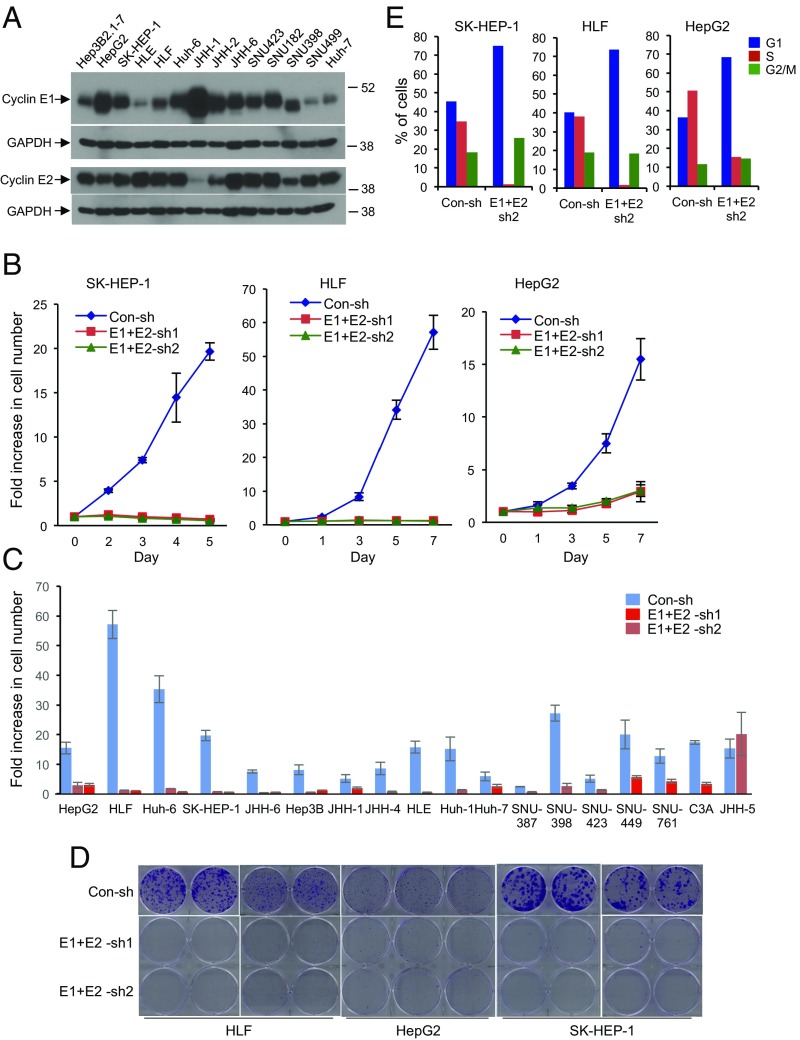
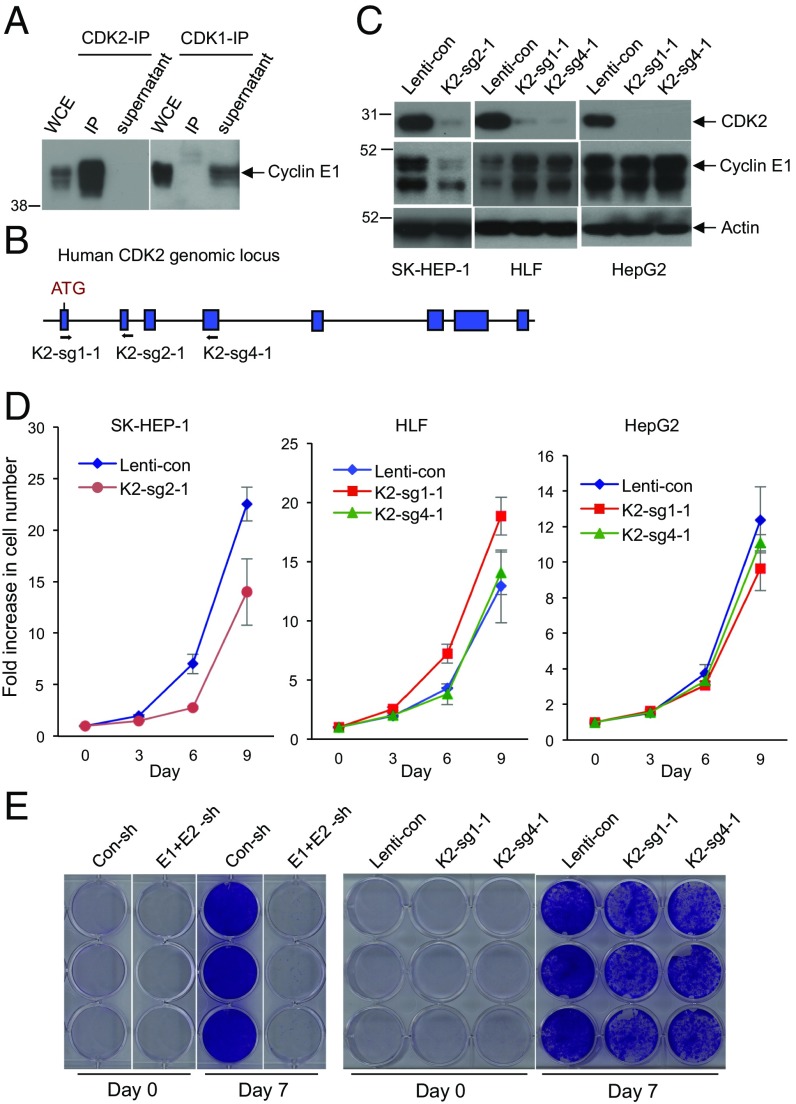
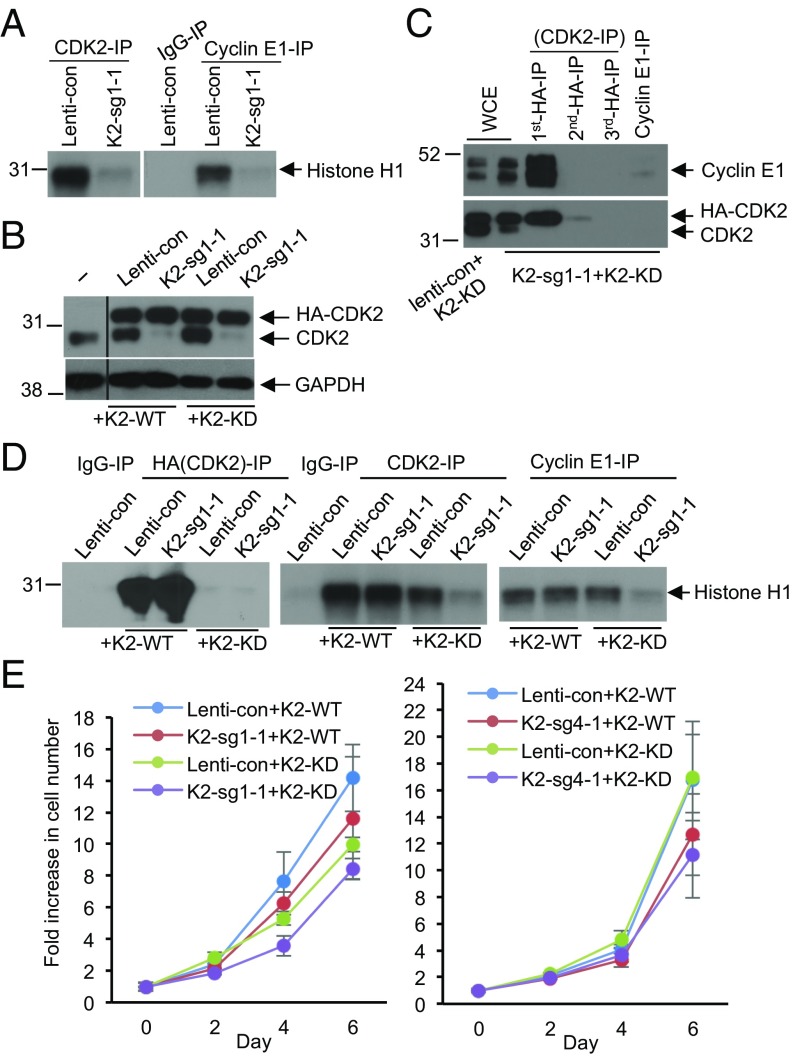
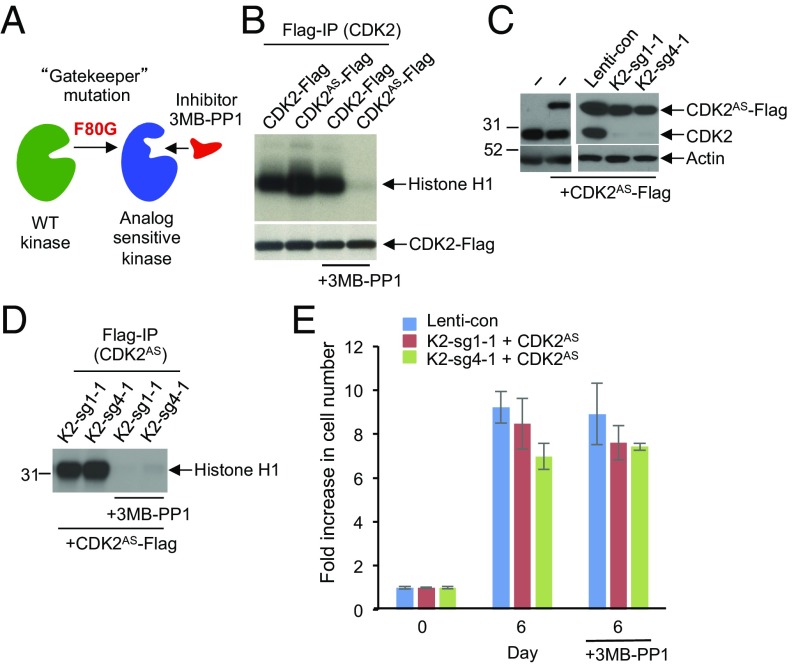
E-type cyclins (cyclins E1 and E2) are components of the core cell cycle machinery and are overexpressed in many human tumor types. E cyclins are thought to drive tumor cell proliferation by activating the cyclin-dependent kinase 2 (CDK2). The cyclin E1 gene represents the site of recurrent integration of the hepatitis B virus in the pathogenesis of hepatocellular carcinoma, and this event is associated with strong up-regulation of cyclin E1 expression. Regardless of the underlying mechanism of tumorigenesis, the majority of liver cancers overexpress E-type cyclins. Here we used conditional cyclin E knockout mice and a liver cancer model to test the requirement for the function of E cyclins in liver tumorigenesis. We show that a ubiquitous, global shutdown of E cyclins did not visibly affect postnatal development or physiology of adult mice. However, an acute ablation of E cyclins halted liver cancer progression. We demonstrated that also human liver cancer cells critically depend on E cyclins for proliferation. In contrast, we found that the function of the cyclin E catalytic partner, CDK2, is dispensable in liver cancer cells. We observed that E cyclins drive proliferation of tumor cells in a CDK2- and kinase-independent mechanism. Our study suggests that compounds which degrade or inhibit cyclin E might represent a highly selective therapeutic strategy for patients with liver cancer, as these compounds would selectively cripple proliferation of tumor cells, while sparing normal tissues.
Keywords: E-type cyclins; cell cycle; cyclin-dependent kinase CDK2; liver cancer.
Conflict of interest statement
Conflict of interest statement: P.S. is a consultant and a recipient of a research grant from Novartis.
Figures
References
-
- Hwang HC, Clurman BE. Cyclin E in normal and neoplastic cell cycles. Oncogene. 2005;24:2776–2786. - PubMed
-
- Gao S, Ma JJ, Lu C. Prognostic value of cyclin E expression in breast cancer: A meta-analysis. Tumour Biol. 2013;34:3423–3430. - PubMed
-
- Patch AM, et al. Australian Ovarian Cancer Study Group Whole-genome characterization of chemoresistant ovarian cancer. Nature. 2015;521:489–494. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
