

Dot1 regulates nucleosome dynamics by its inherent histone chaperone activity in yeast
- PMID: 29339748
- PMCID: PMC5770421
- DOI: 10.1038/s41467-017-02759-8
Dot1 regulates nucleosome dynamics by its inherent histone chaperone activity in yeast
Abstract
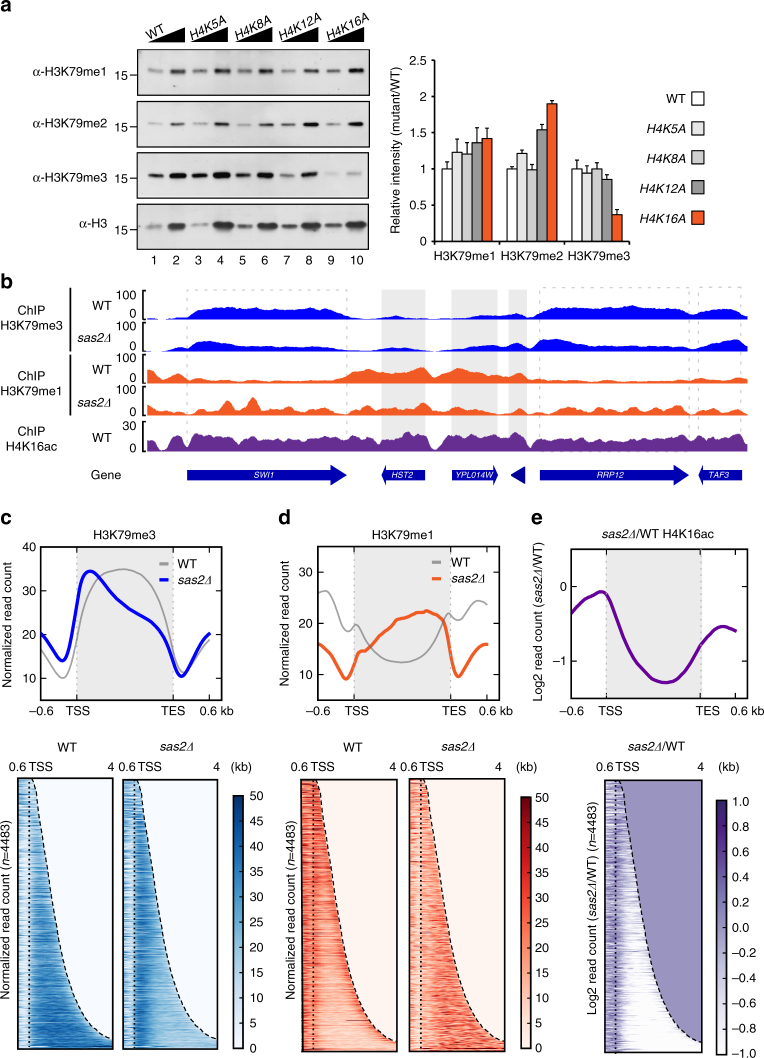
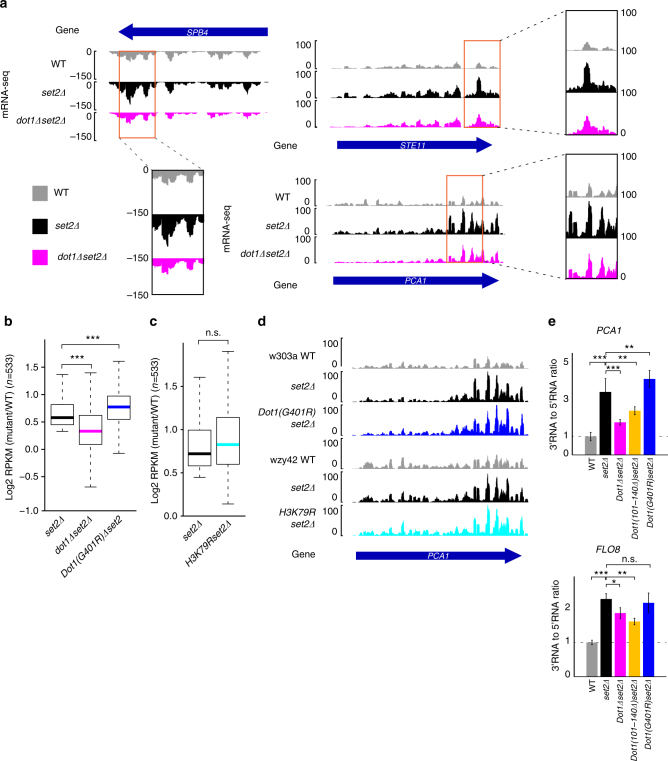
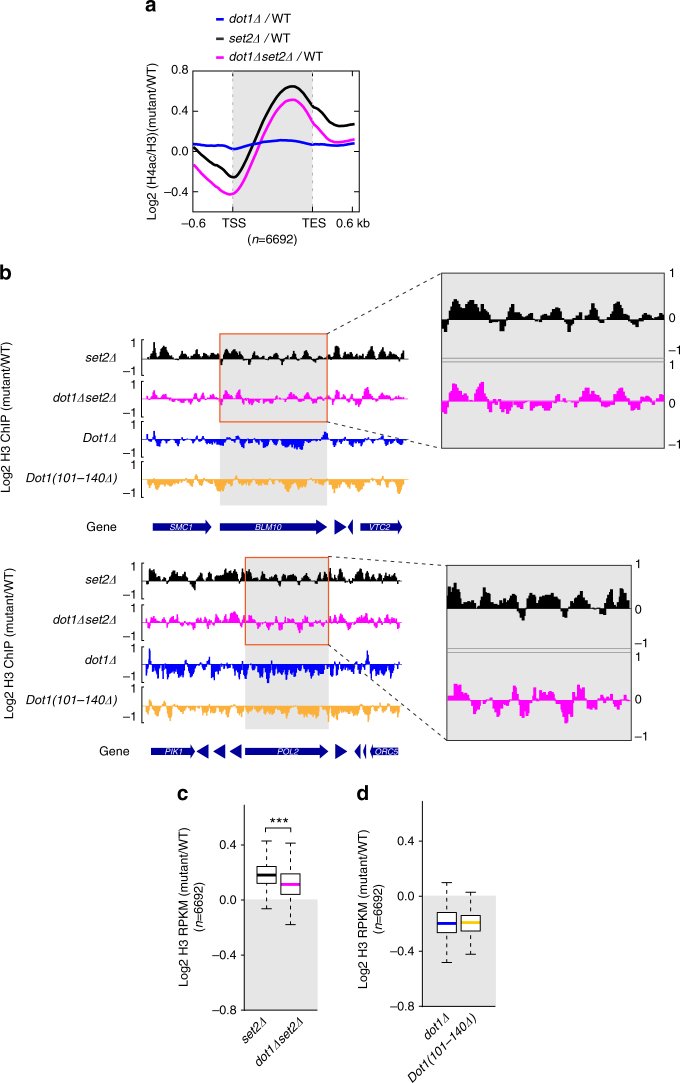
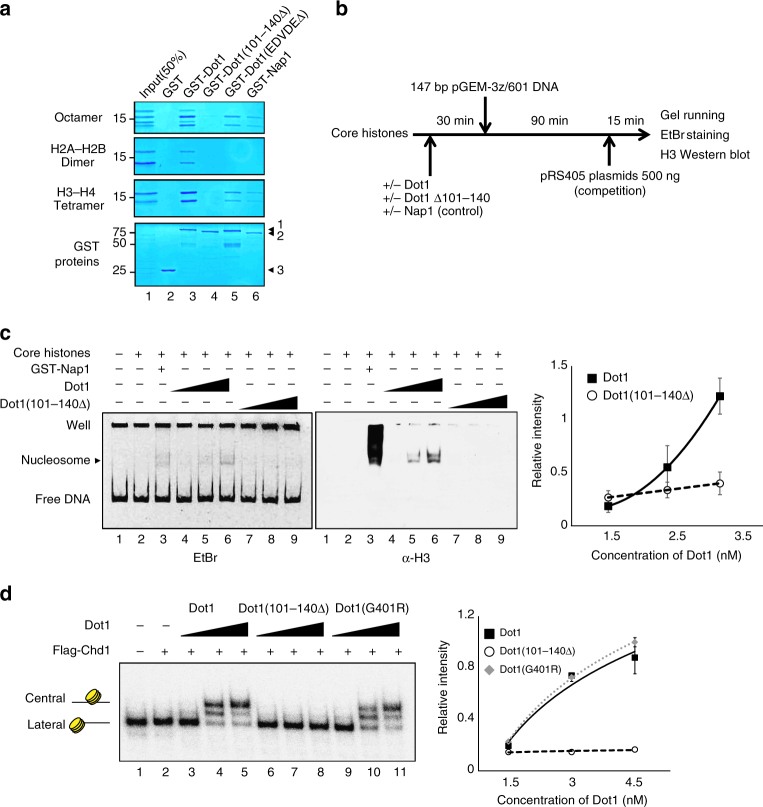
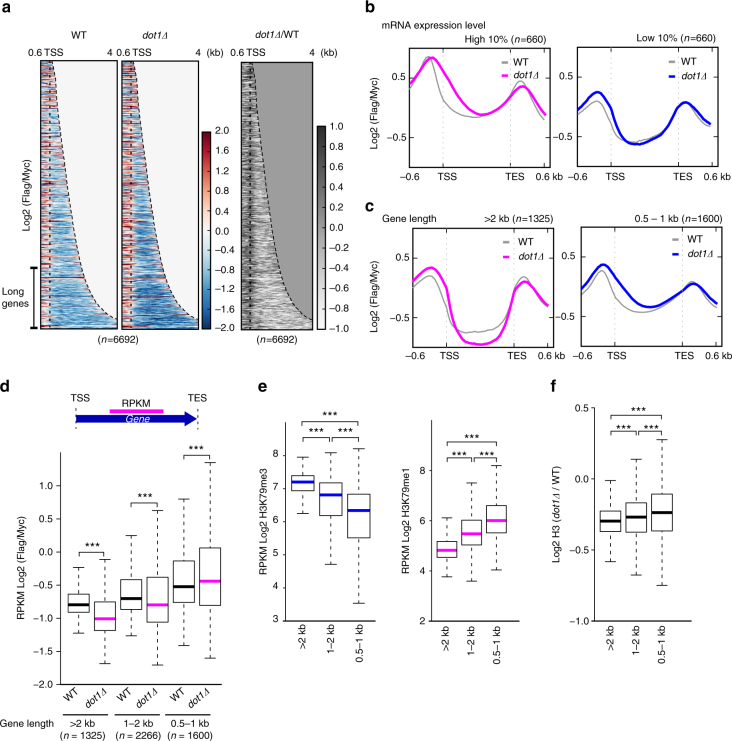
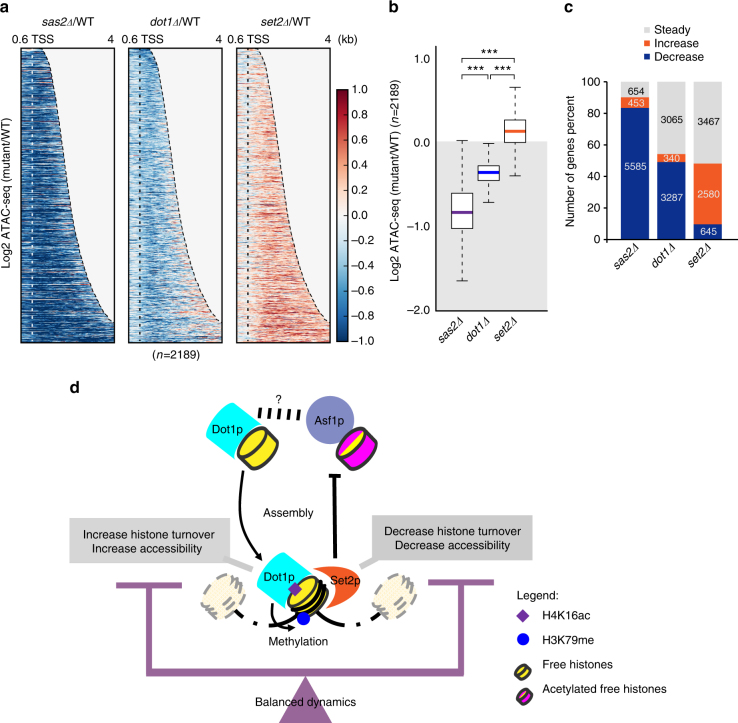
Dot1 (disruptor of telomeric silencing-1, DOT1L in humans) is the only known enzyme responsible for histone H3 lysine 79 methylation (H3K79me) and is evolutionarily conserved in most eukaryotes. Yeast Dot1p lacks a SET domain and does not methylate free histones and thus may have different actions with respect to other histone methyltransferases. Here we show that Dot1p displays histone chaperone activity and regulates nucleosome dynamics via histone exchange in yeast. We show that a methylation-independent function of Dot1p is required for the cryptic transcription within transcribed regions seen following disruption of the Set2-Rpd3S pathway. Dot1p can assemble core histones to nucleosomes and facilitate ATP-dependent chromatin-remodeling activity through its nucleosome-binding domain, in vitro. Global analysis indicates that Dot1p appears to be particularly important for histone exchange and chromatin accessibility on the transcribed regions of long-length genes. Our findings collectively suggest that Dot1p-mediated histone chaperone activity controls nucleosome dynamics in transcribed regions.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
