

Mitochondrial DNA damage and subsequent activation of Z-DNA binding protein 1 links oxidative stress to inflammation in epithelial cells
- PMID: 29343810
- PMCID: PMC5772643
- DOI: 10.1038/s41598-018-19216-1
Mitochondrial DNA damage and subsequent activation of Z-DNA binding protein 1 links oxidative stress to inflammation in epithelial cells
Abstract
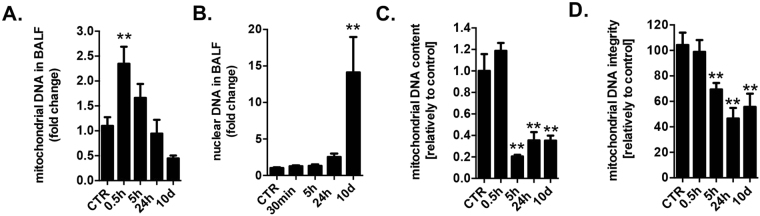
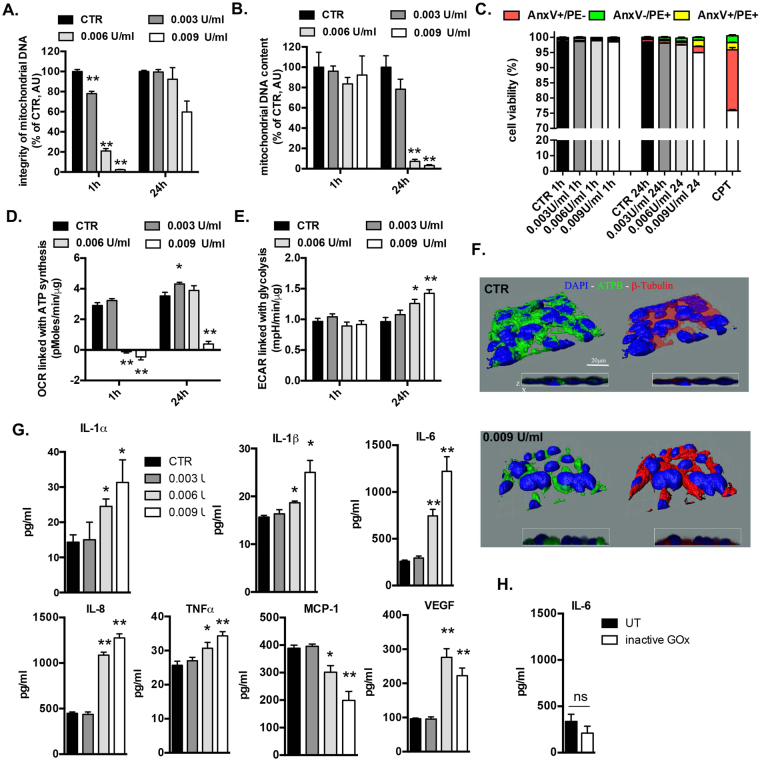
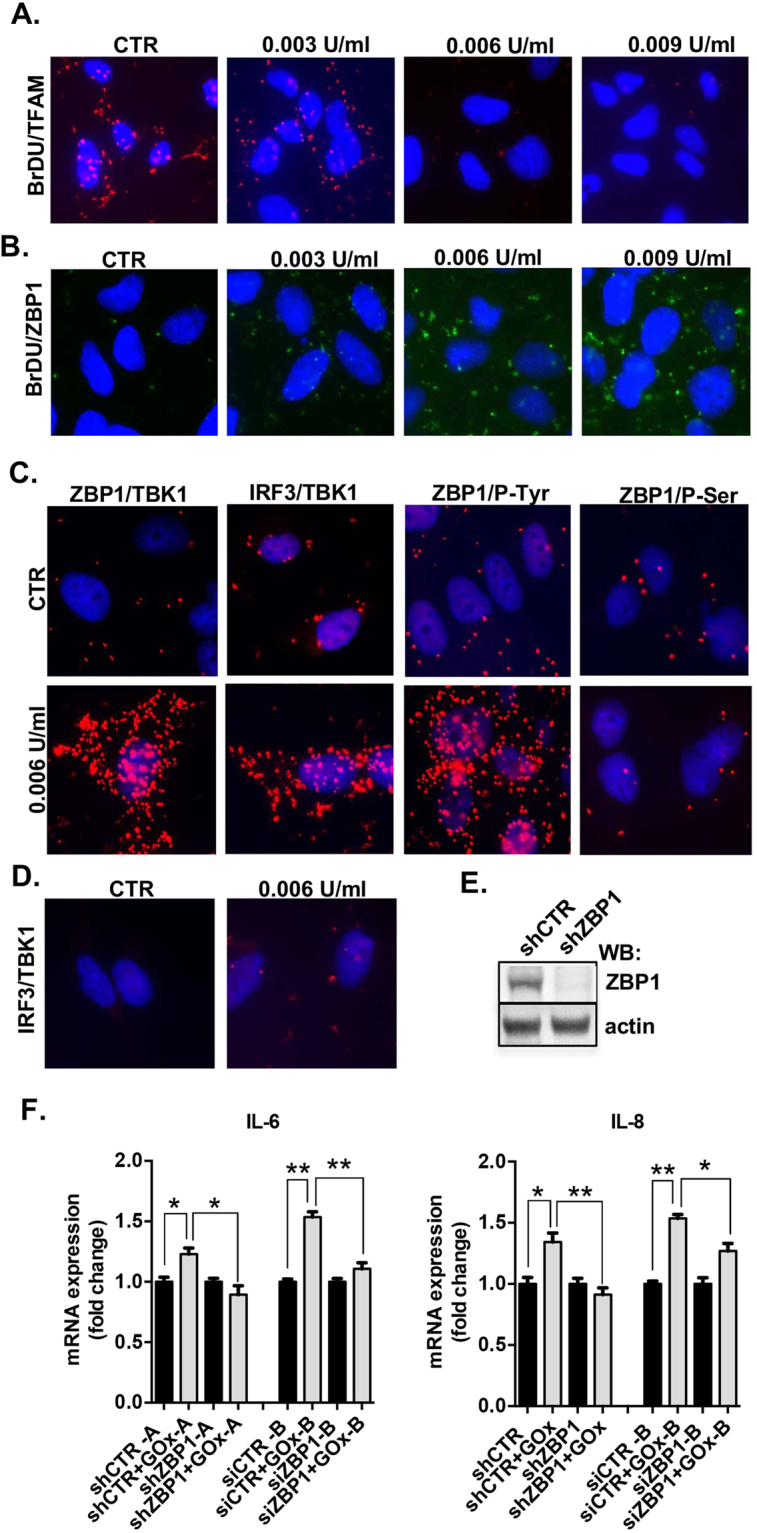
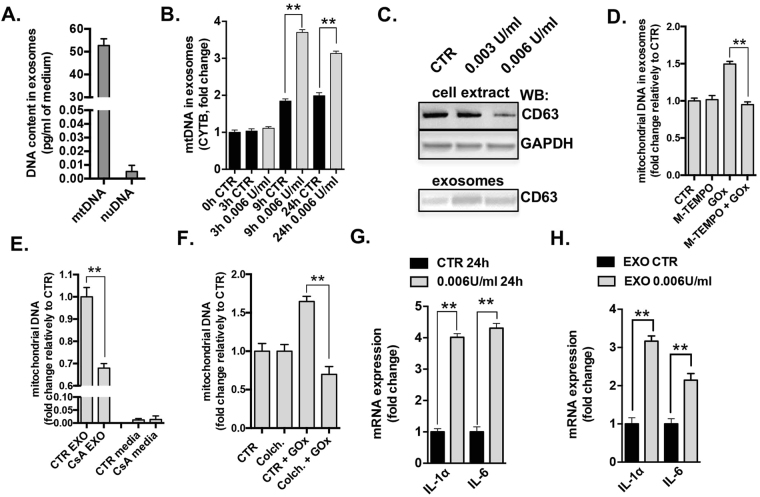
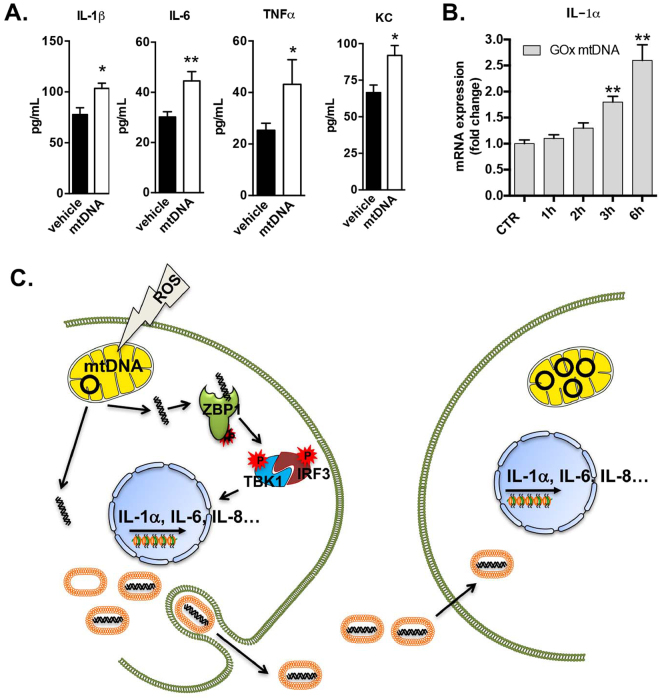
This report identifies mitochondrial DNA (mtDNA) as a target and active mediator that links low-level oxidative stress to inflammatory response in pulmonary epithelial cells. Extrusion of mtDNA into the bronchoalveolar lavage fluid occurs as an early event in mice subjected to cigarette smoke injury, concomitantly with the depletion of mtDNA in the lung tissue. In cultured lung epithelial cells, prolonged, low-level oxidative stress damages the mtDNA, without any detectable damage to the nuclear DNA. In turn, cellular depletion of the mtDNA occurs, together with a transient remodeling of cellular bioenergetics and morphology - all without any detectable impairment in overall cell viability. Damaged mtDNA first enters the cytoplasm, where it binds to Z-DNA binding protein 1 (ZBP1) and triggers inflammation via the TANK-binding kinase 1 /interferon regulatory factor 3 signaling pathway. Fragments of the mtDNA are subsequently released into the extracellular space via exosomes. MtDNA-containing exosomes are capable of inducing an inflammatory response in naïve (non-oxidatively stressed) epithelial cells. In vivo, administration of isolated mtDNA into the in lungs of naïve mice induces the production of pro-inflammatory mediators, without histopathologic evidence of tissue injury. We propose that mtDNA-specific damage, and subsequent activation of the ZBP1 pathway, is a mechanism that links prolonged, low-level oxidative stress to autocrine and paracrine inflammation during the early stages of inflammatory lung disease.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
