

Measuring Entropy in Molecular Recognition by Proteins
- PMID: 29345988
- PMCID: PMC7071556
- DOI: 10.1146/annurev-biophys-060414-034042
Measuring Entropy in Molecular Recognition by Proteins
Abstract
Molecular recognition by proteins is fundamental to the molecular basis of biology. Dissection of the thermodynamic landscape governing protein-ligand interactions has proven difficult because determination of various entropic contributions is quite challenging. Nuclear magnetic resonance relaxation measurements, theory, and simulations suggest that conformational entropy can be accessed through a dynamical proxy. Here, we review the relationship between measures of fast side-chain motion and the underlying conformational entropy. The dynamical proxy reveals that the contribution of conformational entropy can range from highly favorable to highly unfavorable and demonstrates the potential of this key thermodynamic variable to modulate protein-ligand interactions. The dynamical so-called entropy meter also refines the role of solvent entropy and directly determines the loss in rotational-translational entropy that occurs upon formation of high-affinity complexes. The ability to quantify the roles of entropy through an entropy meter based on measurable dynamical properties promises to highlight its role in protein function.
Keywords: NMR relaxation; conformational entropy; molecular recognition; protein dynamics; rotational–translational entropy; solvation entropy.
Figures
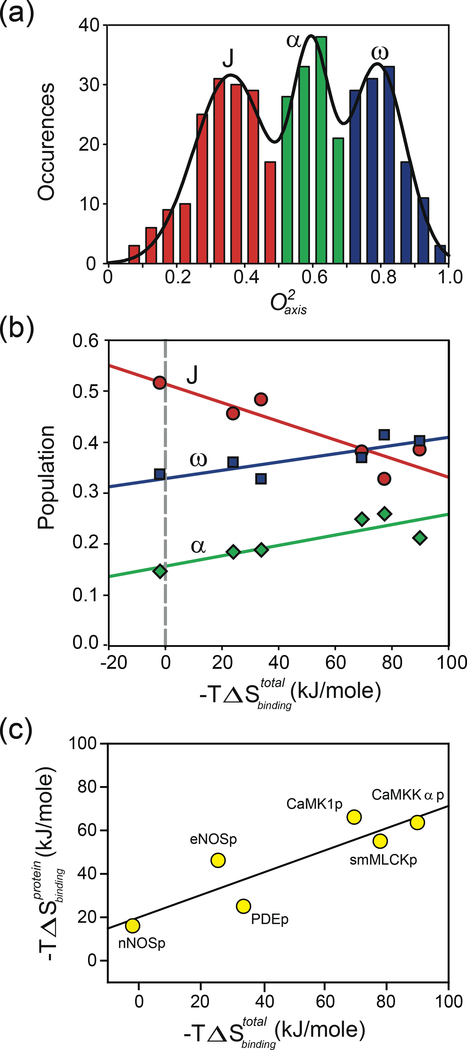
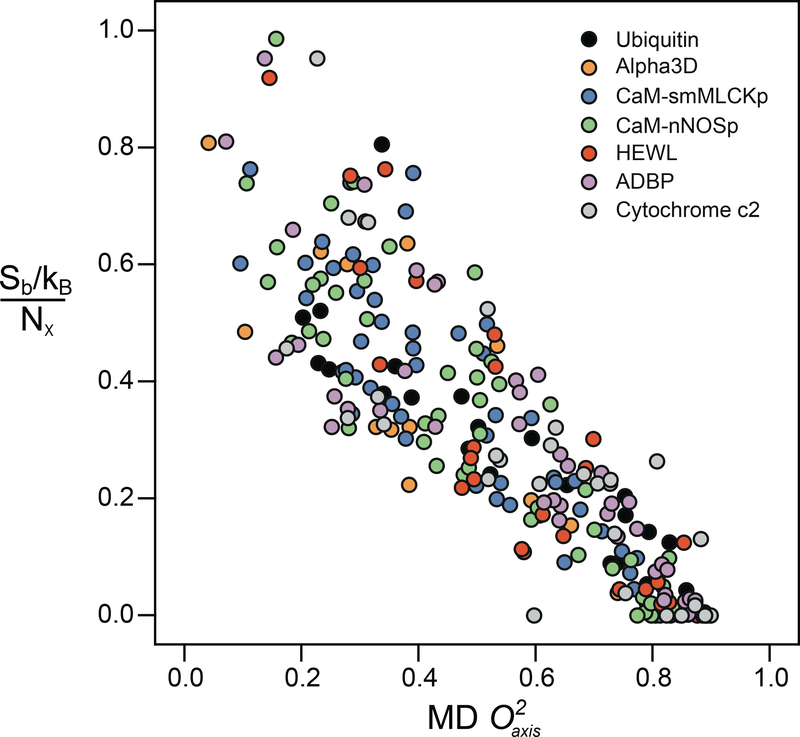
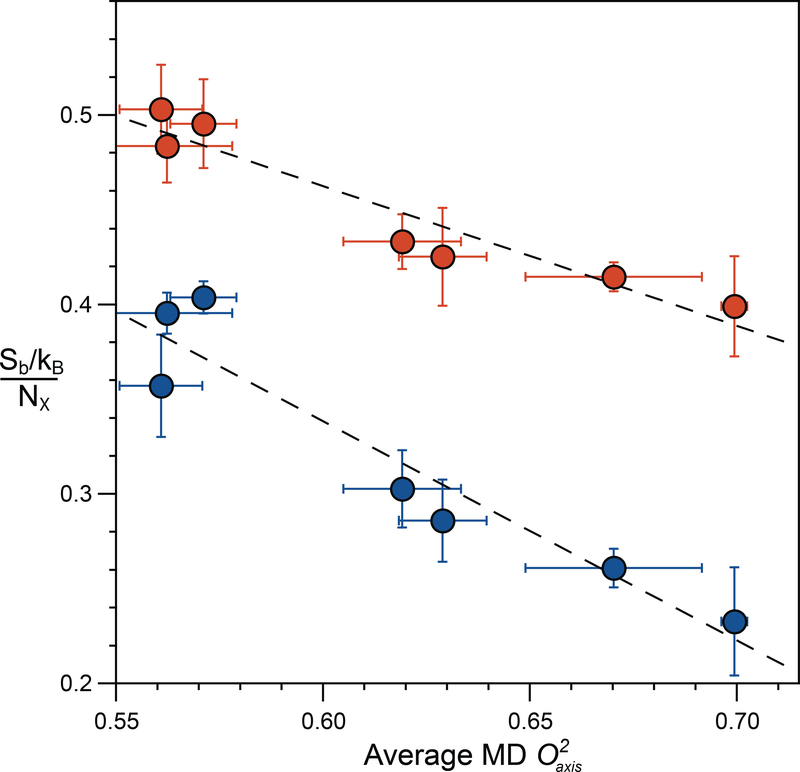
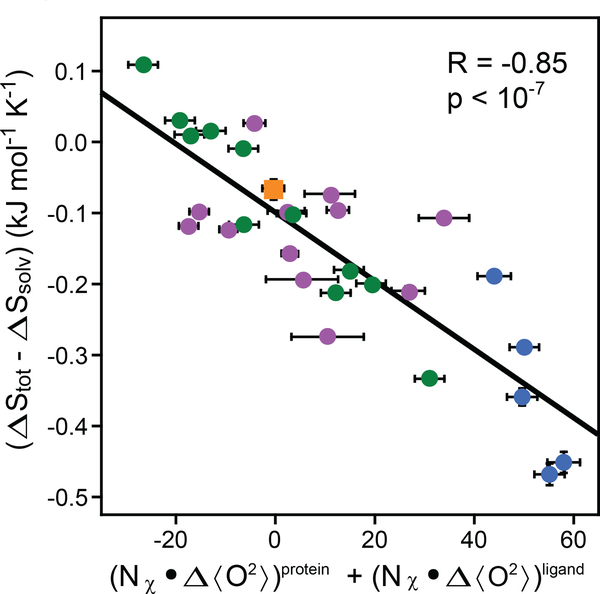
References
-
- Abragam A 1961. Principles of Nuclear Magnetism. Oxford, UK: Clarendon Press
-
-
Akke M, Brüschweiler R, Palmer AG. 1993. NMR order parameters and free energy: an analytical approach and its application to cooperative Ca2+ binding by calbindin-D9k. J. Am. Chem. Soc. 115:9832–33
Used specific potential energy functions to relate motion measurable by NMR relaxation and thermodynamic quantities.
-
-
- Ben-Naim A, Marcus Y. 1984. Solvation thermodynamics of nonionic solutes. J. Chem. Phys. 81:2016–27
-
- Bowman GR. 2015. Accurately modeling nanosecond protein dynamics requires at least microseconds of simulation. J. Comp. Chem. 37:558–66 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
