

Efficacy of MEK inhibition in a K-Ras-driven cholangiocarcinoma preclinical model
- PMID: 29348467
- PMCID: PMC5833851
- DOI: 10.1038/s41419-017-0183-4
Efficacy of MEK inhibition in a K-Ras-driven cholangiocarcinoma preclinical model
Abstract
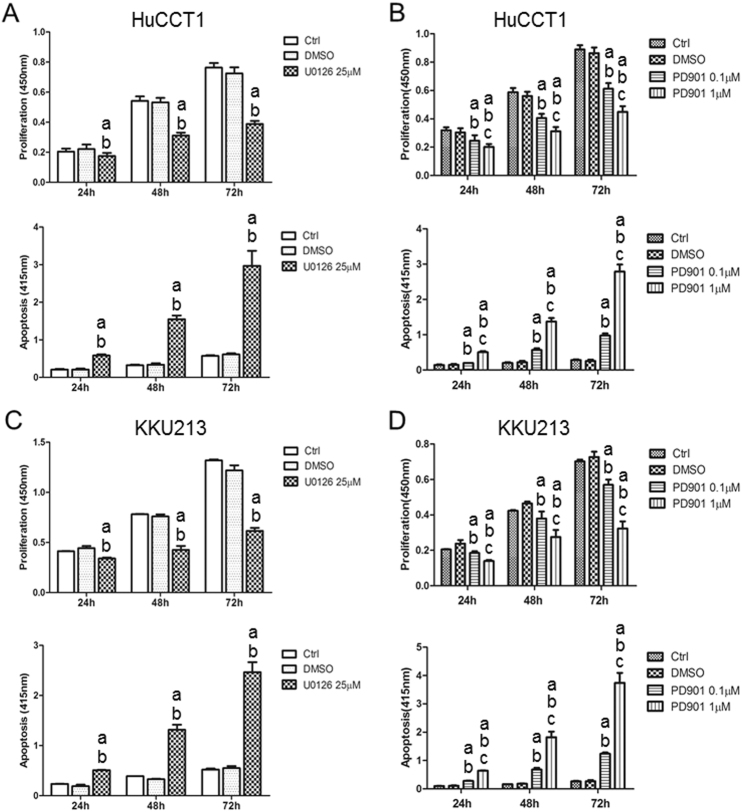
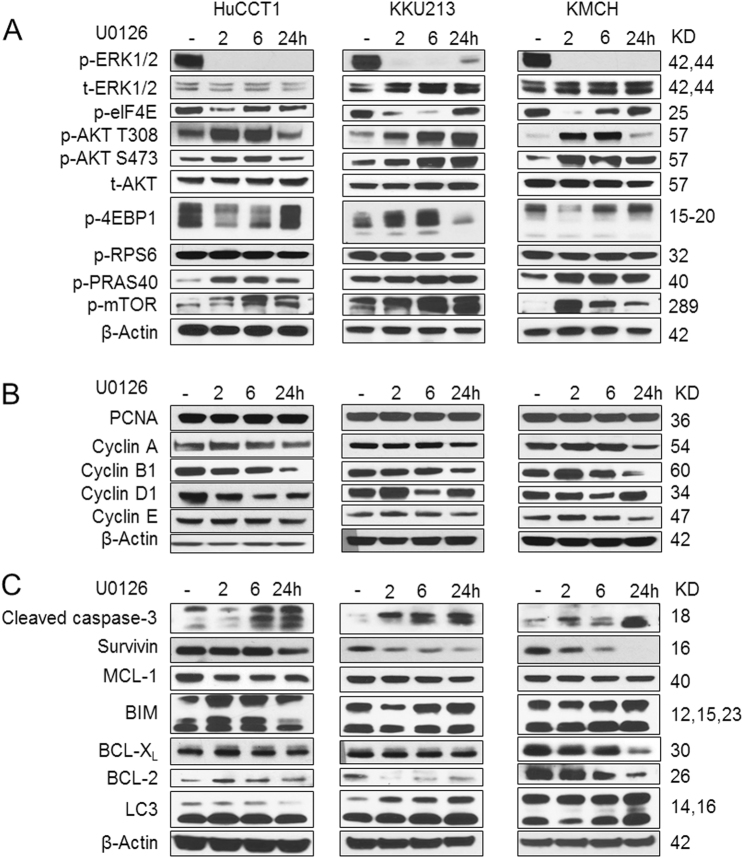
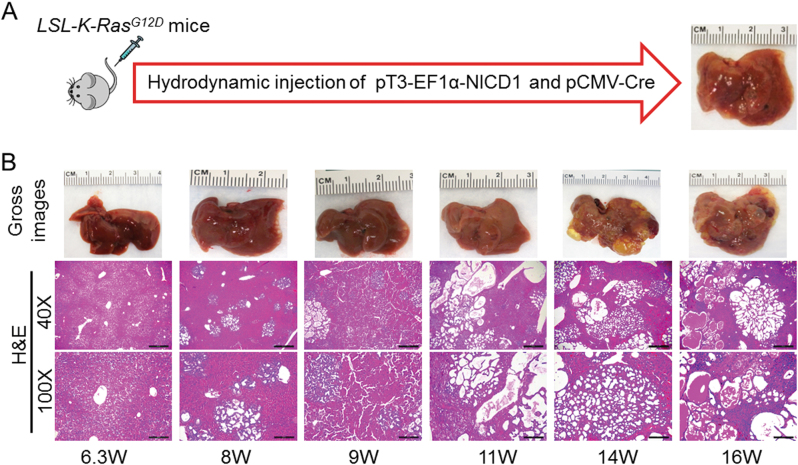
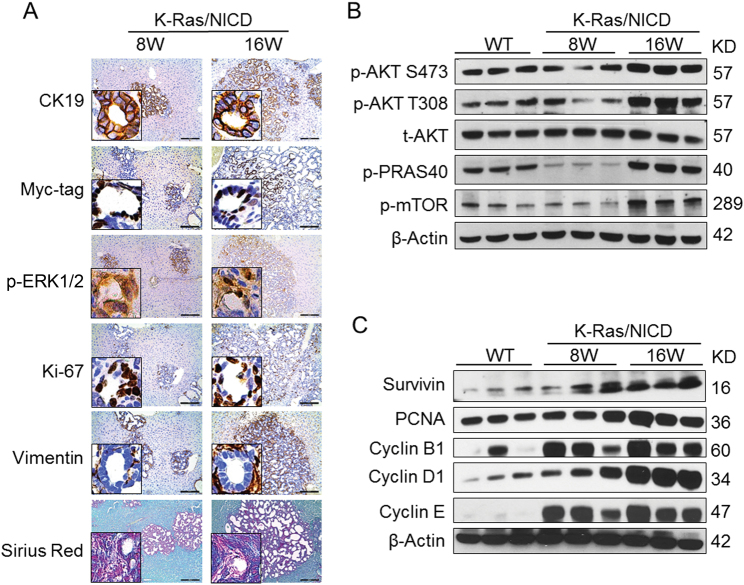
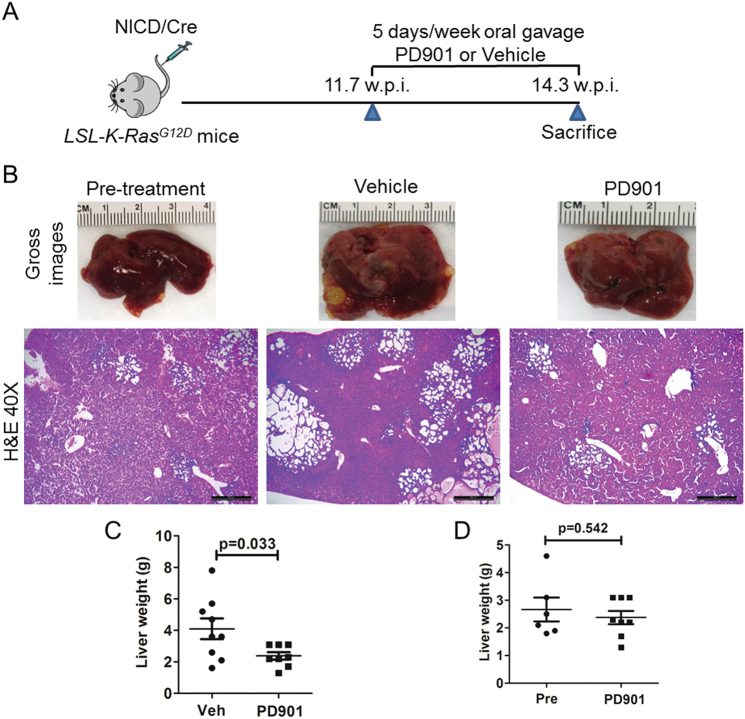
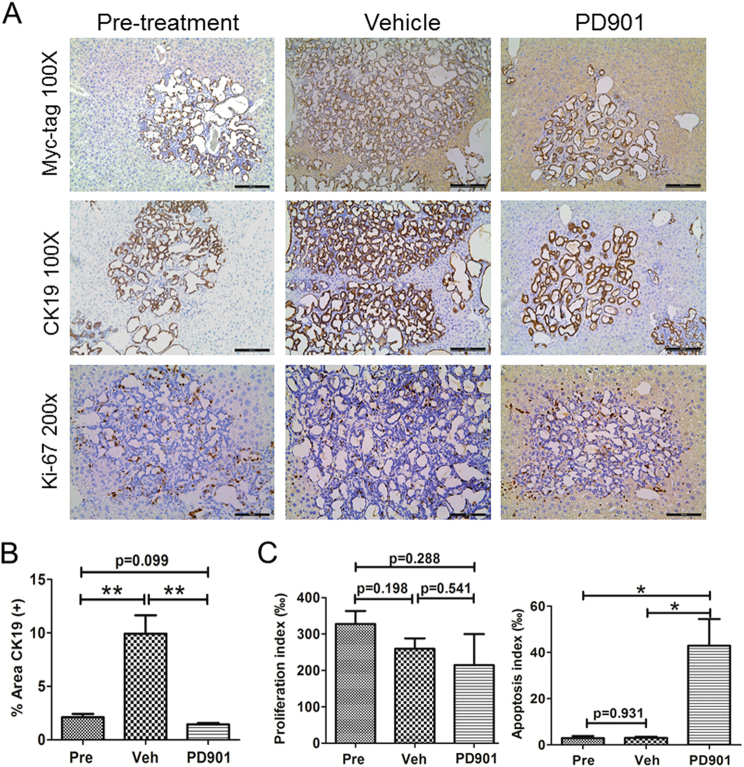
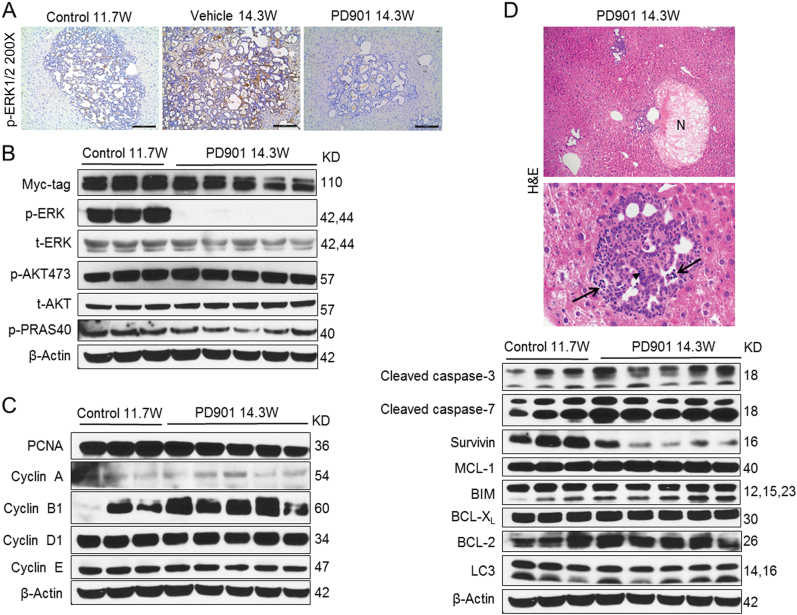
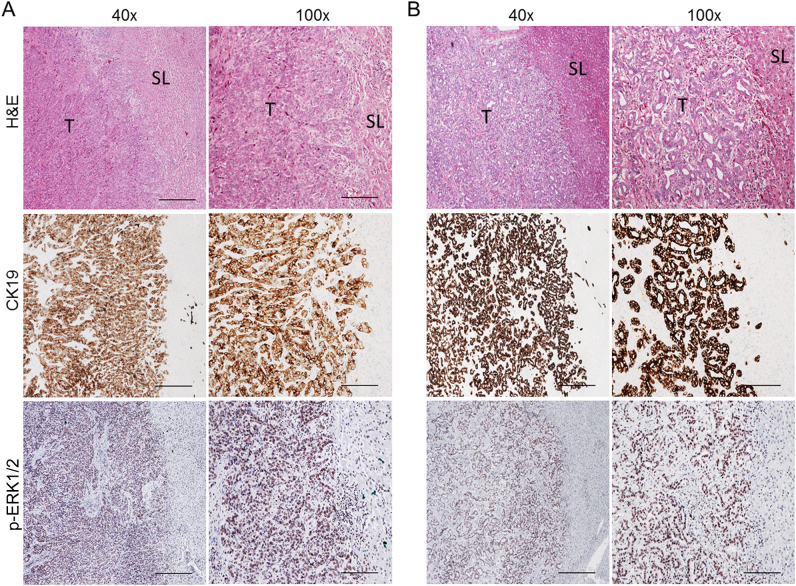
Intrahepatic cholangiocarcinoma (iCCA) is a deadly malignancy with limited treatment options. Gain-of-function mutations in K-Ras is a very frequent alteration, occurring in ~15 to 25% of human iCCA patients. Here, we established a new iCCA model by expressing activated forms of Notch1 (NICD) and K-Ras (K-RasV12D) in the mouse liver (K-Ras/NICD mice). Furthermore, we investigated the therapeutic potential of MEK inhibitors in vitro and in vivo using human CCA cell lines and K-Ras/NICD mice, respectively. Treatment with U0126, PD901, and Selumetinib MEK inhibitors triggered growth restraint in all CCA cell lines tested, with the most pronounced growth suppressive effects being observed in K-Ras mutant cells. Growth inhibition was due to reduction in proliferation and massive apoptosis. Furthermore, treatment of K-Ras/NICD tumor-bearing mice with PD901 resulted in stable disease. At the molecular level, PD901 efficiently inhibited ERK activation in K-Ras/NICD tumor cells, mainly leading to increased apoptosis. Altogether, our study demonstrates that K-Ras/NICD mice represent a novel and useful preclinical model to study K-Ras-driven iCCA development and the effectiveness of MEK inhibitors in counteracting this process. Our data support the usefulness of MEK inhibitors for the treatment of human iCCA.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
