

Emerging potential benefits of modulating NAD+ metabolism in cardiovascular disease
- PMID: 29351465
- PMCID: PMC5966770
- DOI: 10.1152/ajpheart.00409.2017
Emerging potential benefits of modulating NAD+ metabolism in cardiovascular disease
Abstract
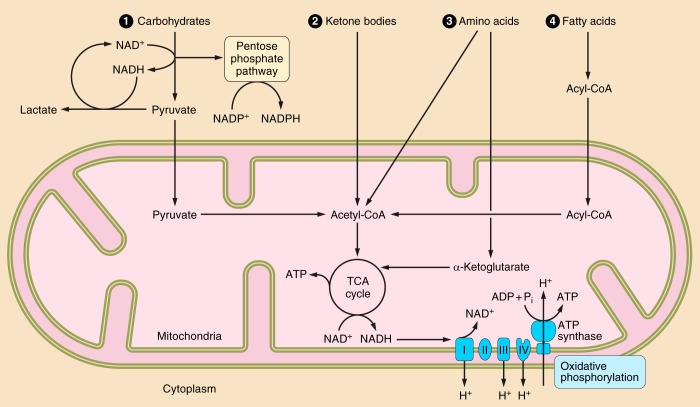
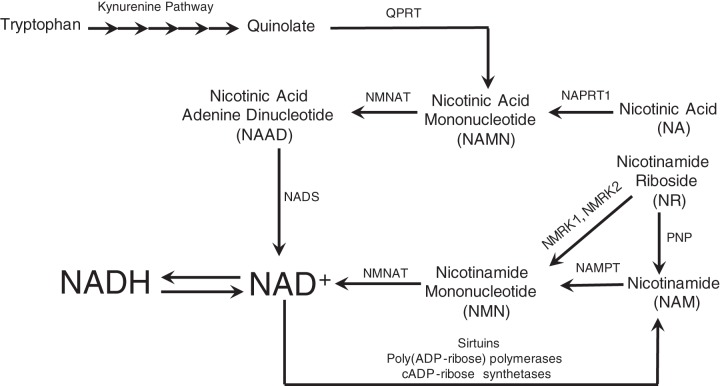
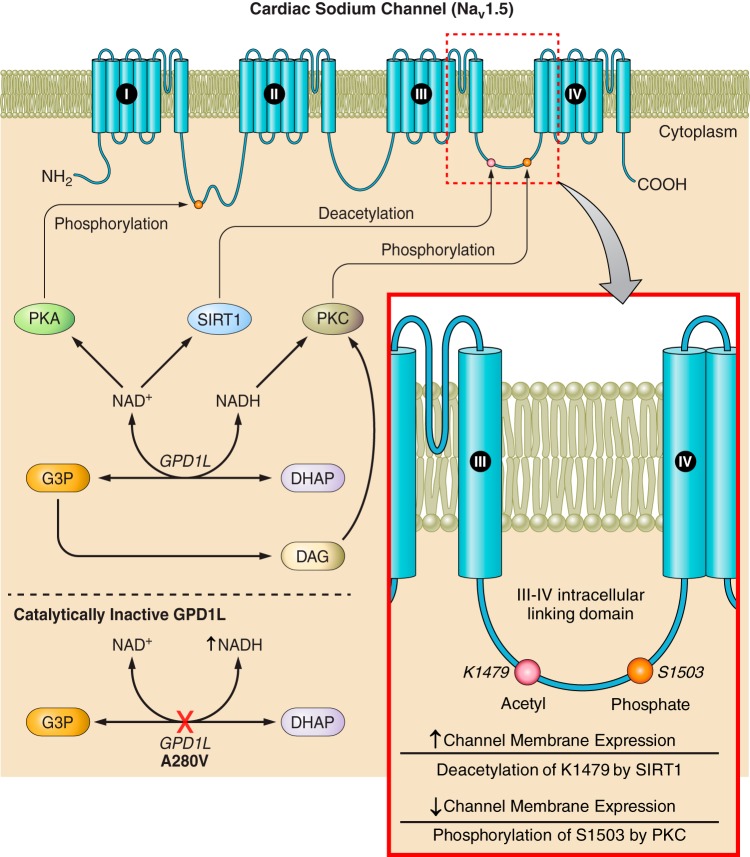
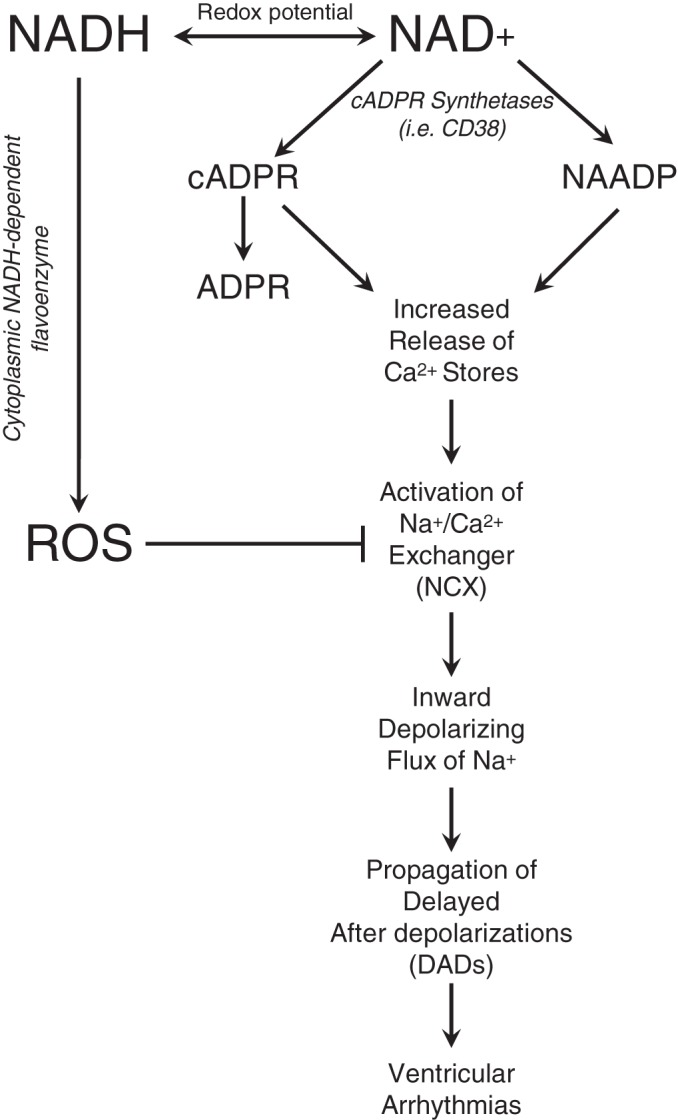
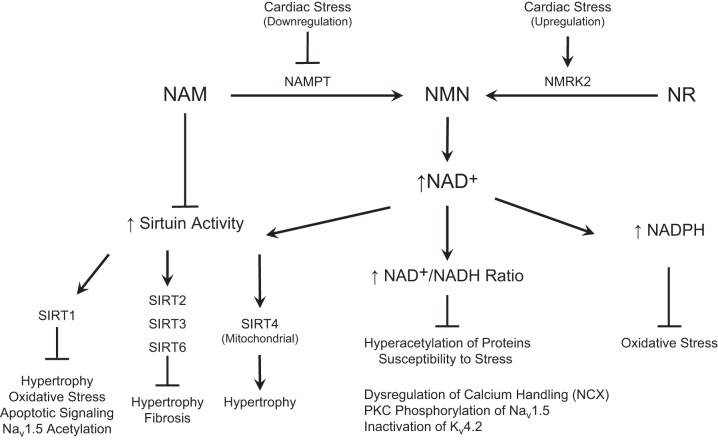
Nicotinamide adenine dinucleotide (NAD+) and related metabolites are central mediators of fuel oxidation and bioenergetics within cardiomyocytes. Additionally, NAD+ is required for the activity of multifunctional enzymes, including sirtuins and poly(ADP-ribose) polymerases that regulate posttranslational modifications, DNA damage responses, and Ca2+ signaling. Recent research has indicated that NAD+ participates in a multitude of processes dysregulated in cardiovascular diseases. Therefore, supplementation of NAD+ precursors, including nicotinamide riboside that boosts or repletes the NAD+ metabolome, may be cardioprotective. This review examines the molecular physiology and preclinical data with respect to NAD+ precursors in heart failure-related cardiac remodeling, ischemic-reperfusion injury, and arrhythmias. In addition, alternative NAD+-boosting strategies and potential systemic effects of NAD+ supplementation with implications on cardiovascular health and disease are surveyed.
Keywords: cardiovascular diseases; ischemia-reperfusion; nicotinamide adenine dinucleotide; oxidation-reduction (redox).
Figures
References
-
- Airhart SE, Shireman LM, Risler LJ, Anderson GD, Nagana Gowda GA, Raftery D, Tian R, Shen DD, O’Brien KD. An open-label, non-randomized study of the pharmacokinetics of the nutritional supplement nicotinamide riboside (NR) and its effects on blood NAD+ levels in healthy volunteers. PLoS One 12: e0186459, 2017. doi:10.1371/journal.pone.0186459. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous