

Determination of protein oligomeric structure from small-angle X-ray scattering
- PMID: 29352739
- PMCID: PMC5866936
- DOI: 10.1002/pro.3376
Determination of protein oligomeric structure from small-angle X-ray scattering
Abstract
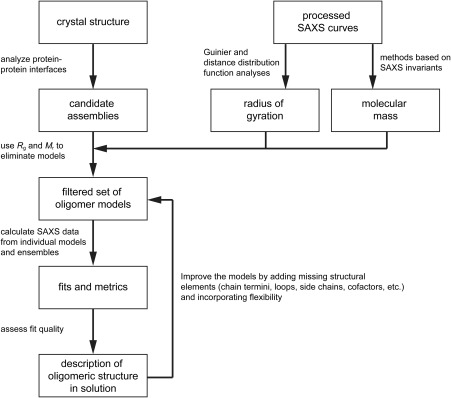
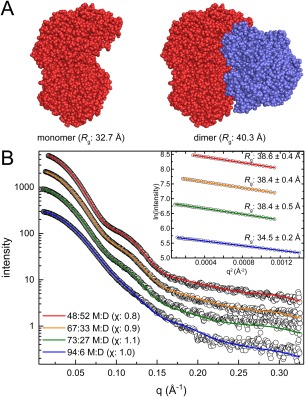
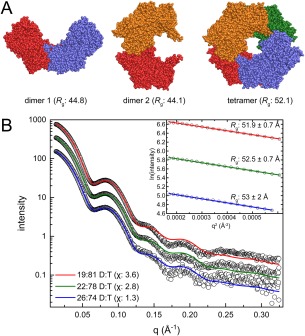
Small-angle X-ray scattering (SAXS) is useful for determining the oligomeric states and quaternary structures of proteins in solution. The average molecular mass in solution can be calculated directly from a single SAXS curve collected on an arbitrary scale from a sample of unknown protein concentration without the need for beamline calibration or protein standards. The quaternary structure in solution can be deduced by comparing the experimental SAXS curve to theoretical curves calculated from proposed models of the oligomer. This approach is especially robust when the crystal structure of the target protein is known, and the candidate oligomer models are derived from the crystal lattice. When SAXS data are obtained at multiple protein concentrations, this analysis can provide insight into dynamic self-association equilibria. Herein, we summarize the computational methods that are used to determine protein molecular mass and quaternary structure from SAXS data. These methods are organized into a workflow and demonstrated with four case studies using experimental SAXS data from the published literature.
Keywords: molecular mass; oligomerization; protein structure; quaternary structure; small-angle X-ray scattering.
© 2018 The Protein Society.
Figures
References
-
- Krissinel E, Henrick K, Detection of protein assemblies in crystals. Computational Life Sciences: First International Symposium. In: Berthold MR, Glen R, Diederichs K, Kohlbacher O, Fischer I, Eds. (2005) Konstanz, Germany: Springer, pp 163–174.
-
- Krissinel E, Henrick K (2007) Inference of macromolecular assemblies from crystalline state. J Mol Biol 372:774–797. - PubMed
-
- Scharer MA, Grutter MG, Capitani G (2010) CRK: an evolutionary approach for distinguishing biologically relevant interfaces from crystal contacts. Proteins 78:2707–2713. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
