

A HYBRID THREE-SCALE MODEL OF TUMOR GROWTH
- PMID: 29353950
- PMCID: PMC5773147
- DOI: 10.1142/S0218202518500021
A HYBRID THREE-SCALE MODEL OF TUMOR GROWTH
Abstract
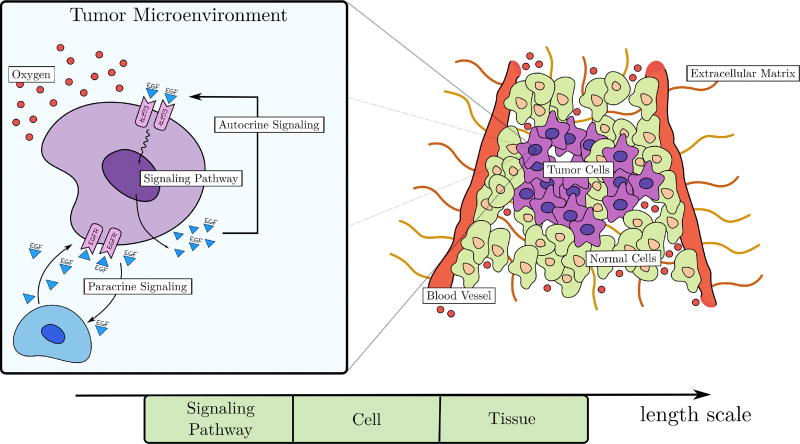
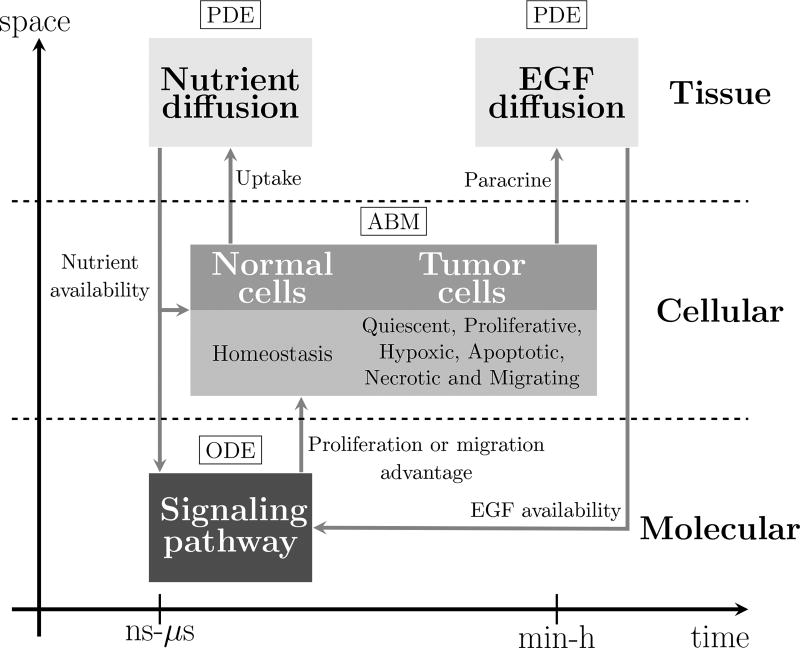
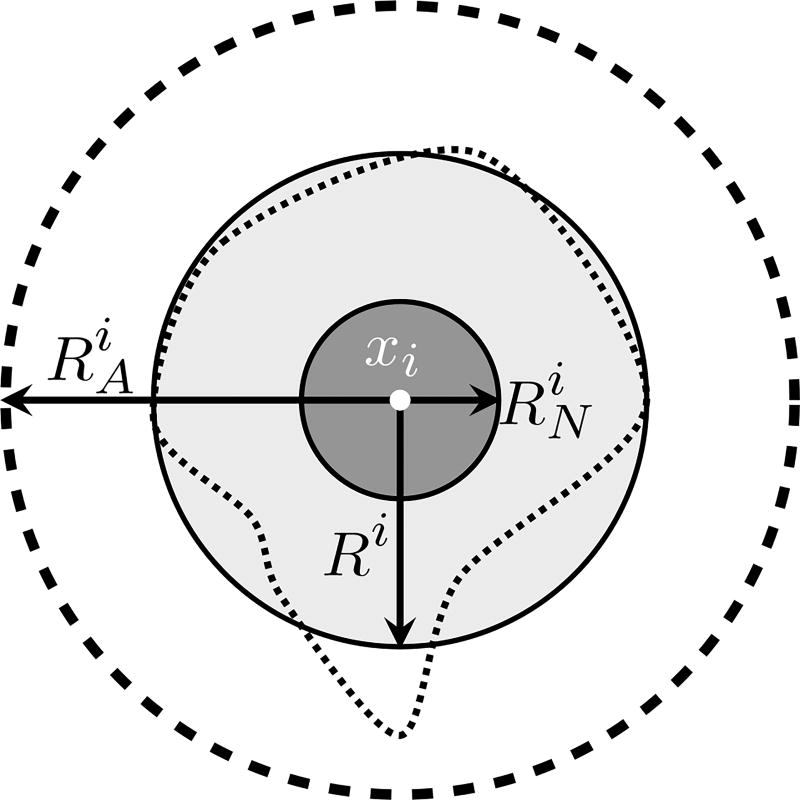
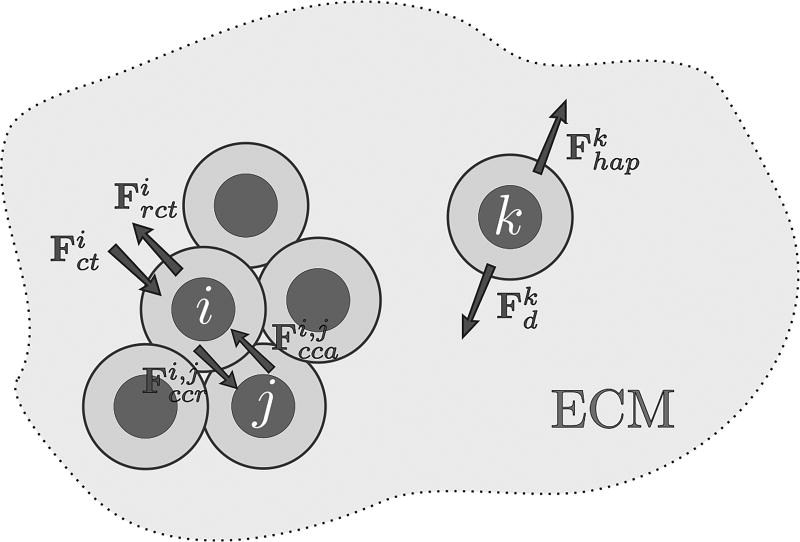
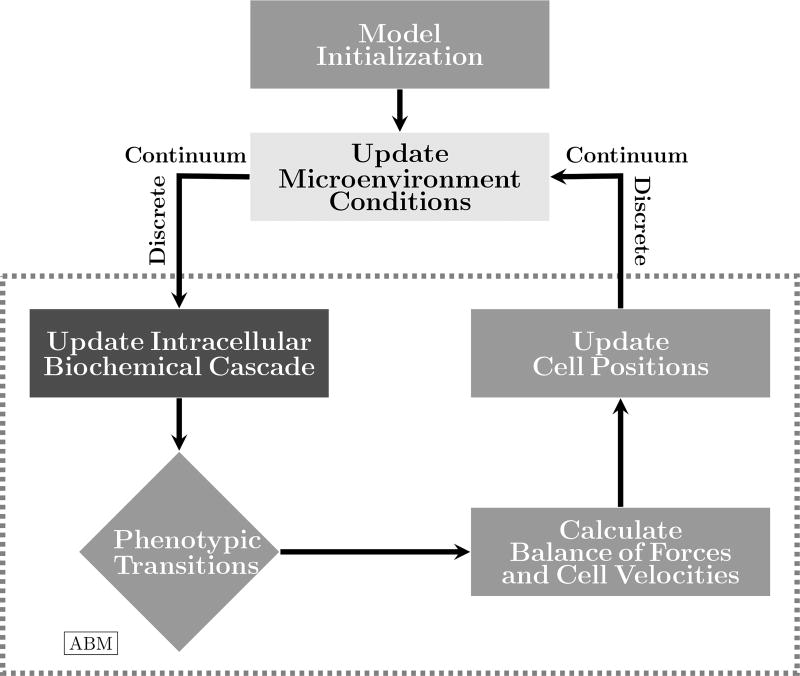
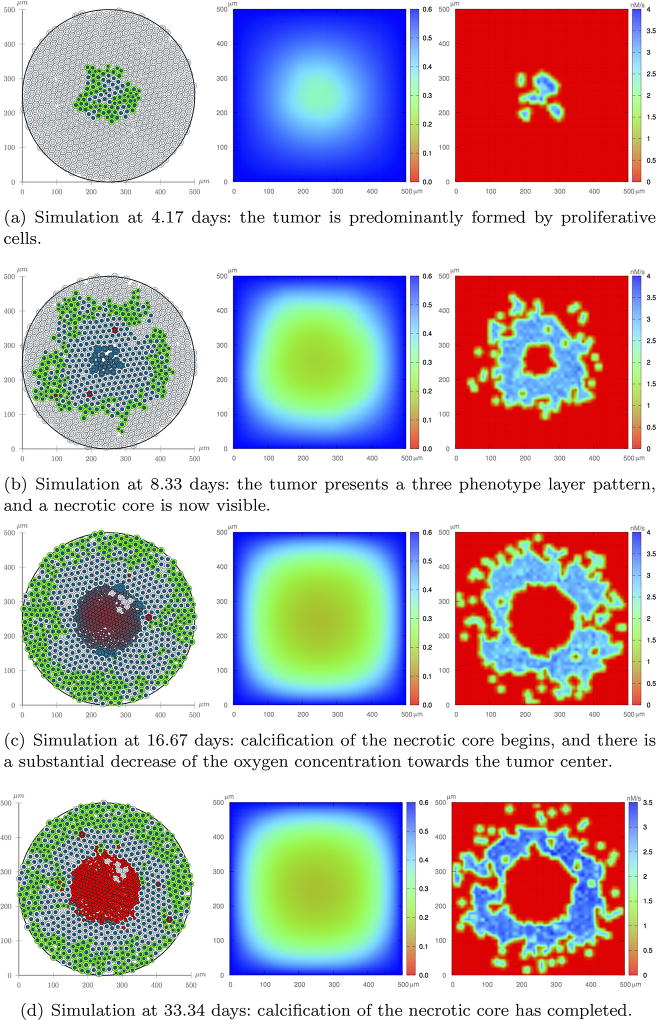
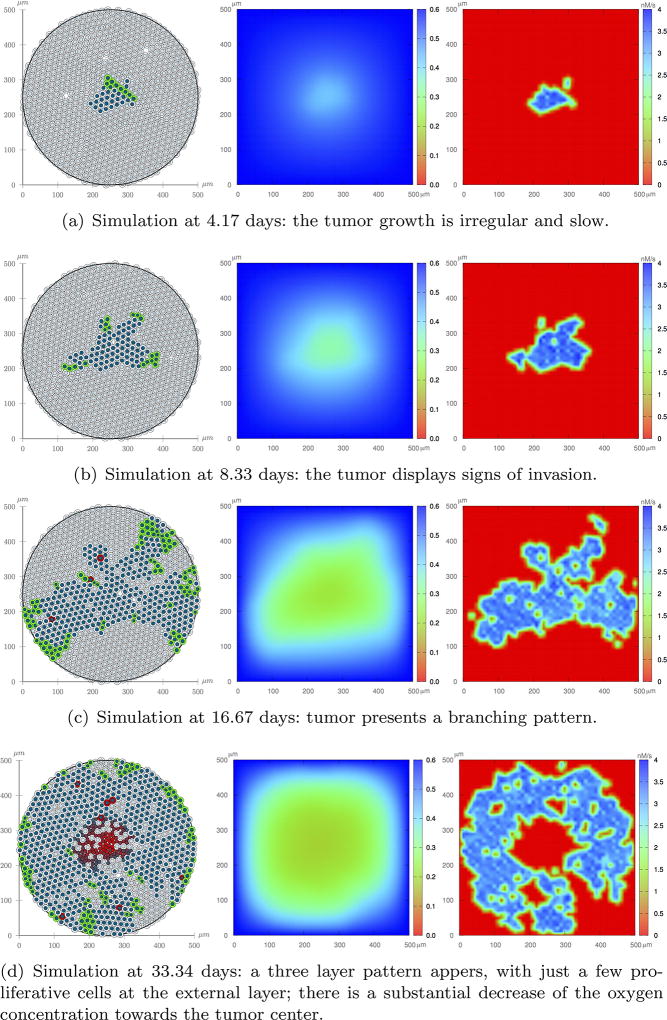
Cancer results from a complex interplay of different biological, chemical, and physical phenomena that span a wide range of time and length scales. Computational modeling may help to unfold the role of multiple evolving factors that exist and interact in the tumor microenvironment. Understanding these complex multiscale interactions is a crucial step towards predicting cancer growth and in developing effective therapies. We integrate different modeling approaches in a multiscale, avascular, hybrid tumor growth model encompassing tissue, cell, and sub-cell scales. At the tissue level, we consider the dispersion of nutrients and growth factors in the tumor microenvironment, which are modeled through reaction-diffusion equations. At the cell level, we use an agent based model (ABM) to describe normal and tumor cell dynamics, with normal cells kept in homeostasis and cancer cells differentiated apoptotic, hypoxic, and necrotic states. Cell movement is driven by the balance of a variety of forces according to Newton's second law, including those related to growth-induced stresses. Phenotypic transitions are defined by specific rule of behaviors that depend on microenvironment stimuli. We integrate in each cell/agent a branch of the epidermal growth factor receptor (EGFR) pathway. This pathway is modeled by a system of coupled nonlinear differential equations involving the mass laws of 20 molecules. The rates of change in the concentration of some key molecules trigger proliferation or migration advantage response. The bridge between cell and tissue scales is built through the reaction and source terms of the partial differential equations. Our hybrid model is built in a modular way, enabling the investigation of the role of different mechanisms at multiple scales on tumor progression. This strategy allows representating both the collective behavior due to cell assembly as well as microscopic intracellular phenomena described by signal transduction pathways. Here, we investigate the impact of some mechanisms associated with sustained proliferation on cancer progression. Specifically, we focus on the intracellular proliferation/migration-advantage-response driven by the EGFR pathway and on proliferation inhibition due to accumulation of growth-induced stresses. Simulations demonstrate that the model can adequately describe some complex mechanisms of tumor dynamics, including growth arrest in avascular tumors. Both the sub-cell model and growth-induced stresses give rise to heterogeneity in the tumor expansion and a rich variety of tumor behaviors.
Keywords: Cell-agent based model; Hybrid multiscale model; Signaling pathway.
Figures
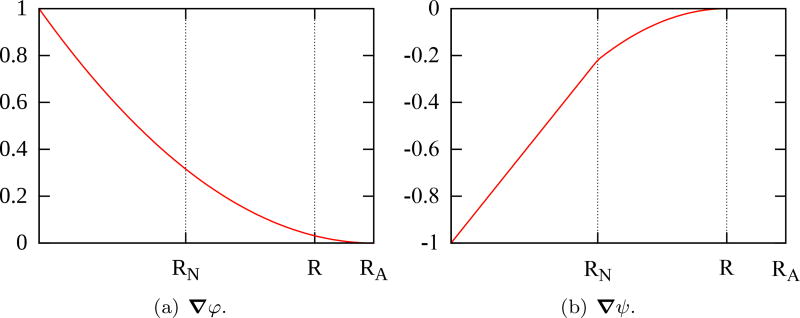
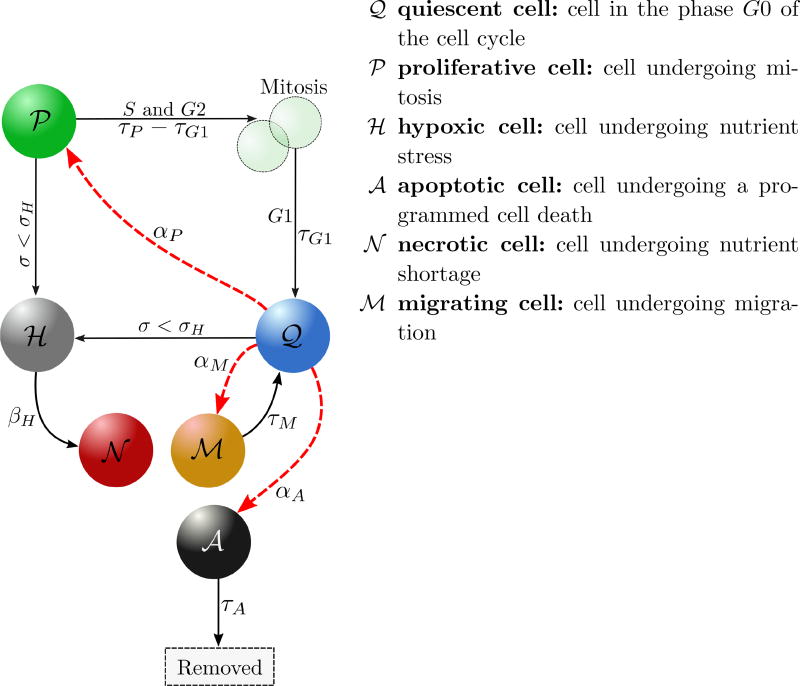
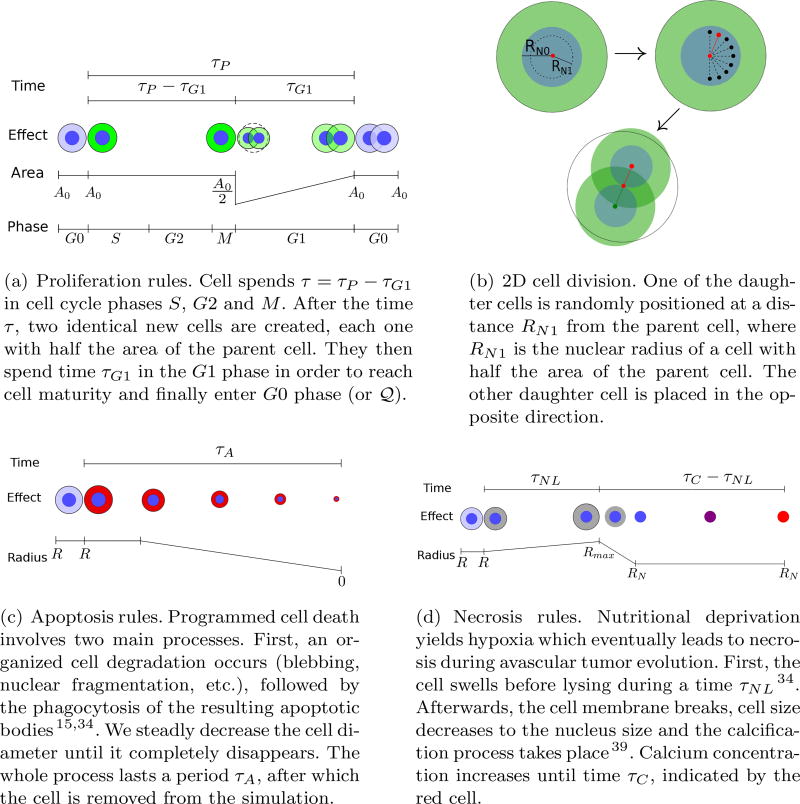
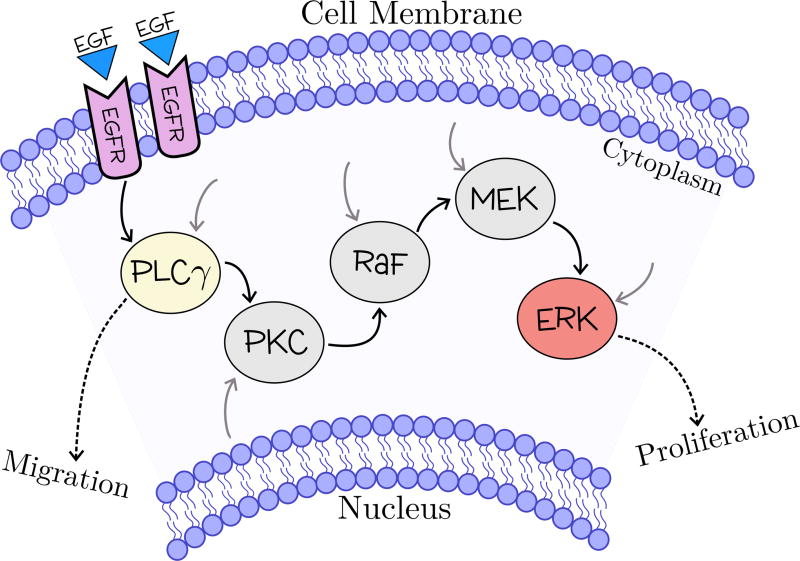

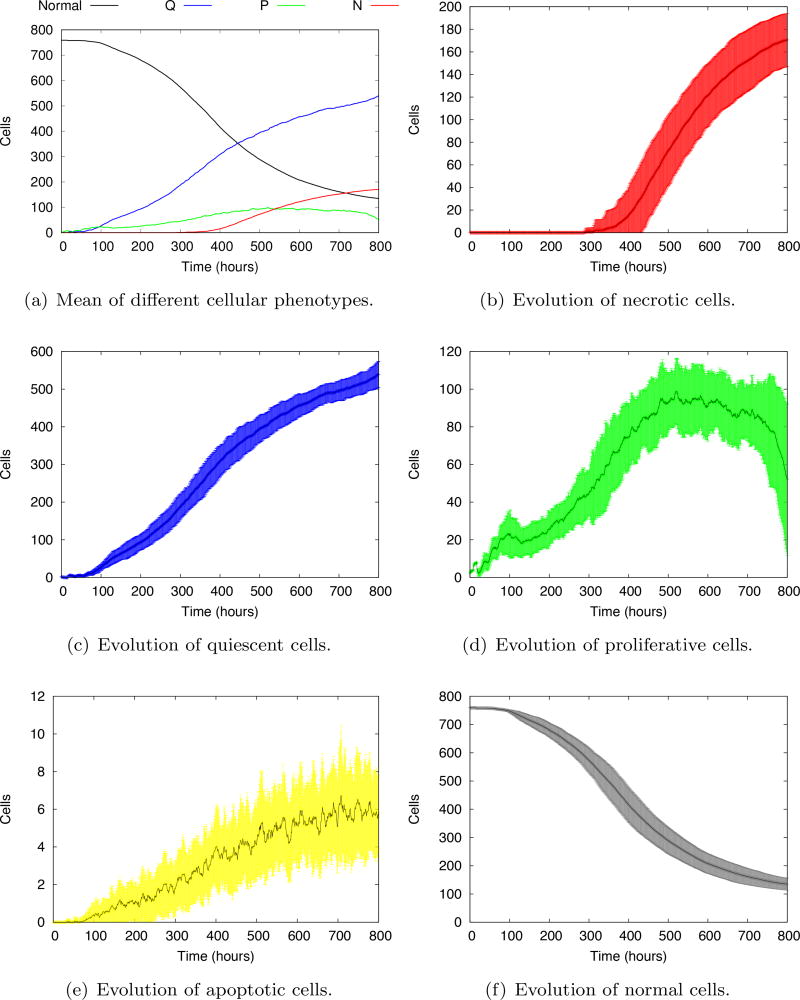
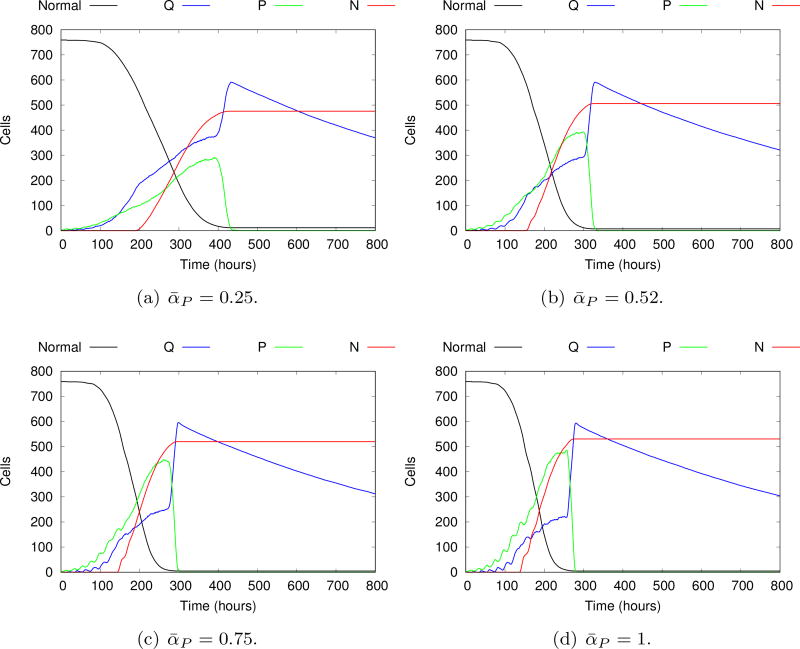
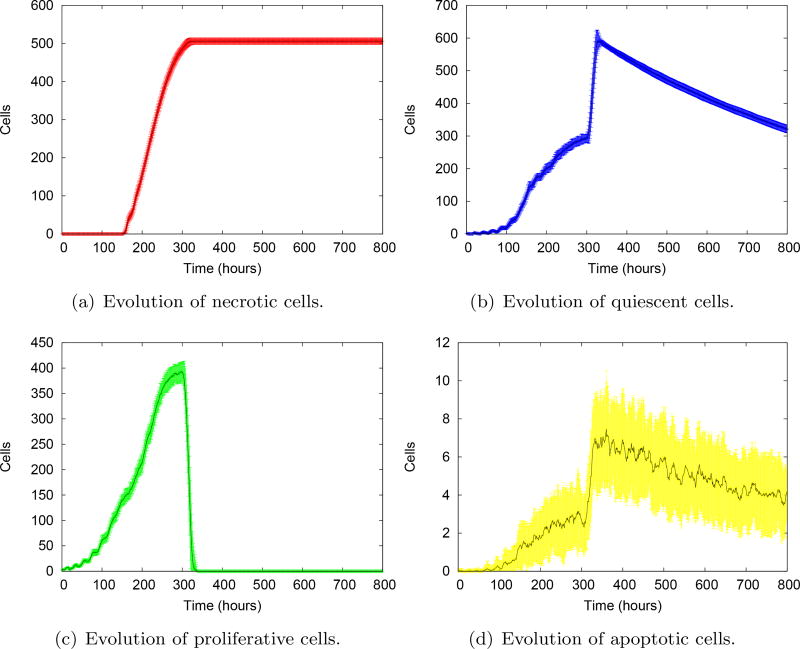
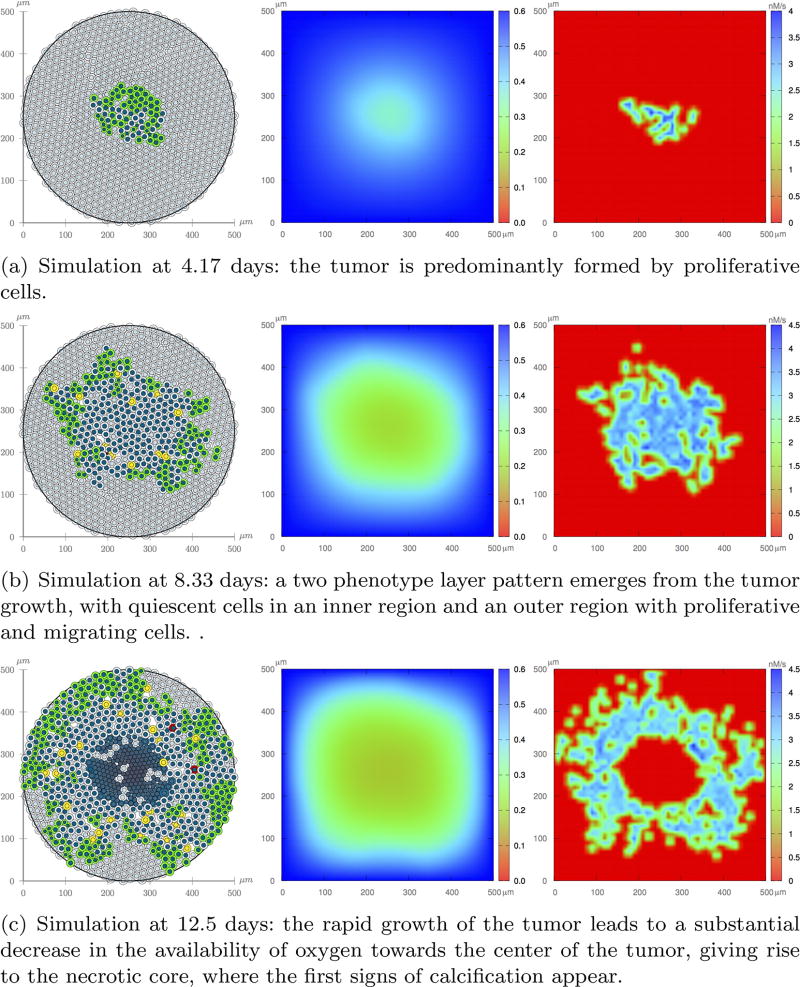
References
-
- Anderson ARA. A hybrid multiscale model of solid tumour growth and invasion: evolution and the microenvironment. chapter I.1. Birkháuser; 2007. pp. 3–28.
-
- Anderson Alexander RA, Chaplain Mark AJ, Rejniak Katarzyna A. Single-cell-based models in biology and medicine. Birkhäuser Verlag AG; 2007.
-
- Athale Chaitanya, Deisboeck Thomas S. The effects of egf-receptor density on multiscale tumor growth patterns. Journal of Theoretical Biology. 2006;238:771–779. - PubMed
-
- Athale Chaitanya, Mansury Yuri, Deisboeck Thomas S. Simulating the impact of a molecular “decision-process” on cellular phenotype and multicellular patterns in brain tumors. Journal of Theoretical Biology. 2005;233:469–481. - PubMed
-
- Byrne HM. Mathematics and Life Sciences. chapter 12. University of Oxford; 2012. pp. 279–312.
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous