

Zn2+-induced disruption of neuronal mitochondrial function: Synergism with Ca2+, critical dependence upon cytosolic Zn2+ buffering, and contributions to neuronal injury
- PMID: 29355498
- PMCID: PMC5849513
- DOI: 10.1016/j.expneurol.2018.01.012
Zn2+-induced disruption of neuronal mitochondrial function: Synergism with Ca2+, critical dependence upon cytosolic Zn2+ buffering, and contributions to neuronal injury
Abstract
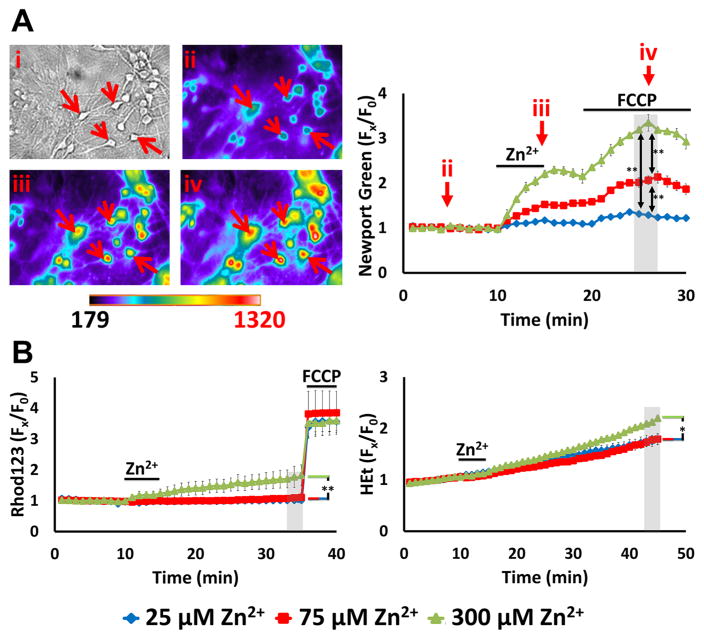
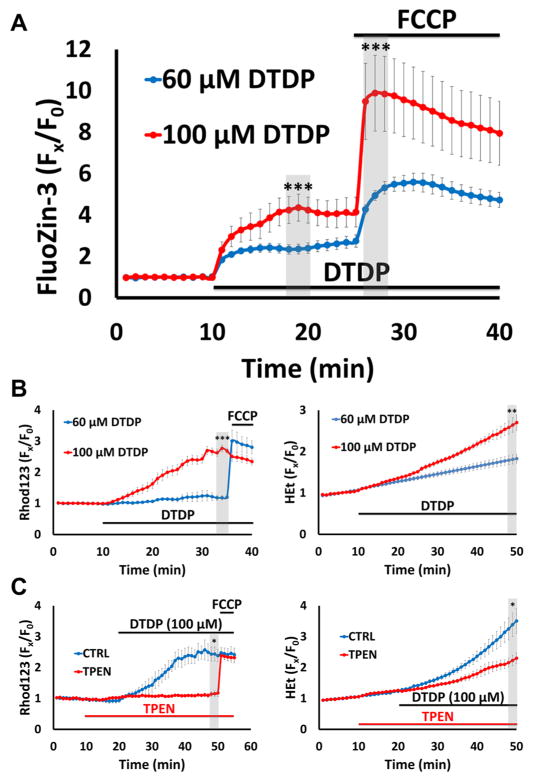
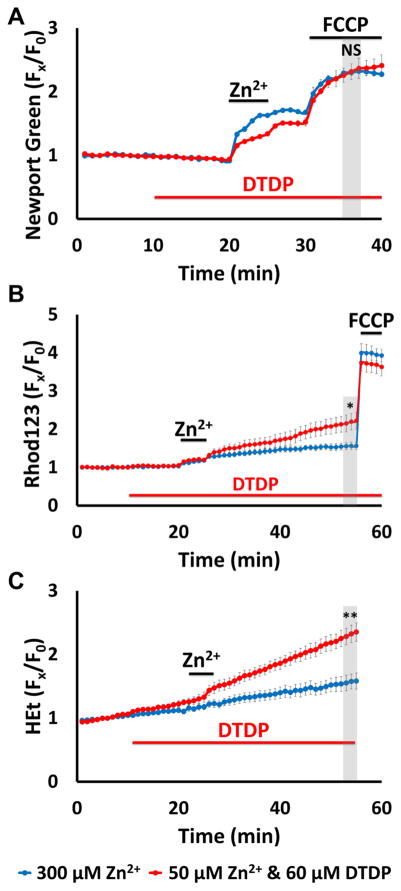
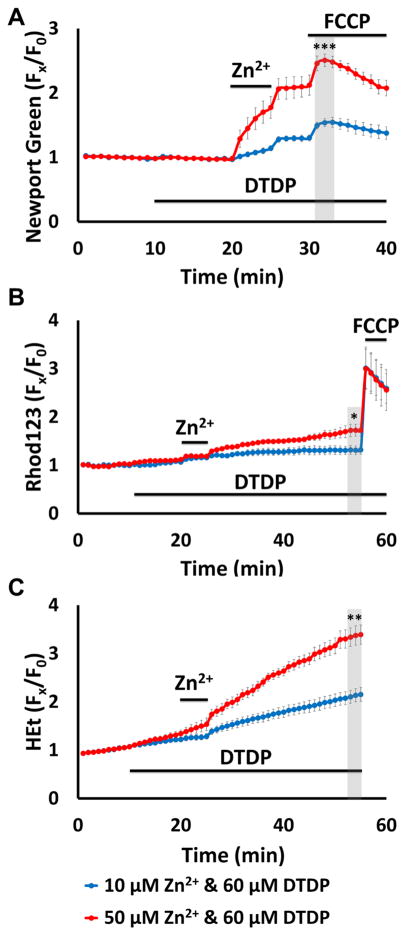
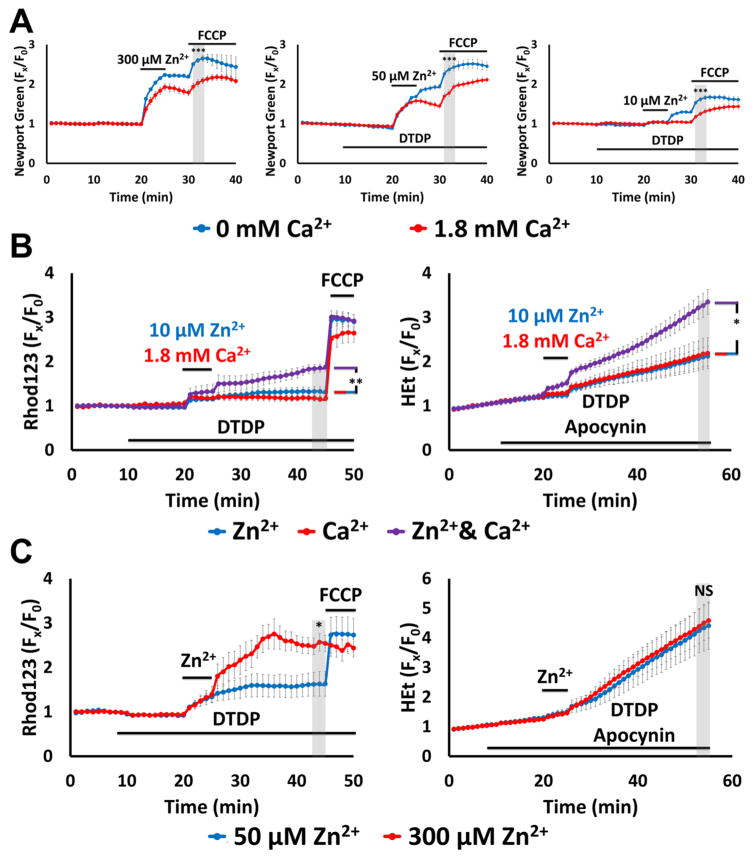
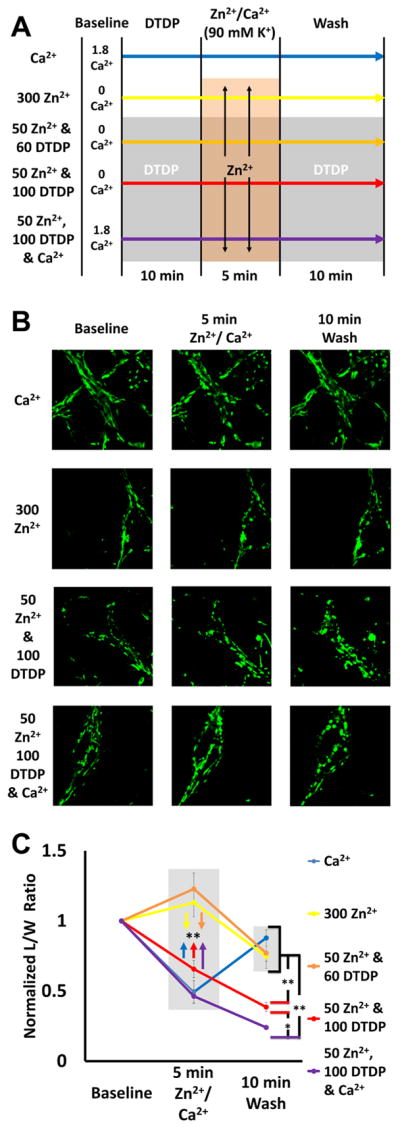
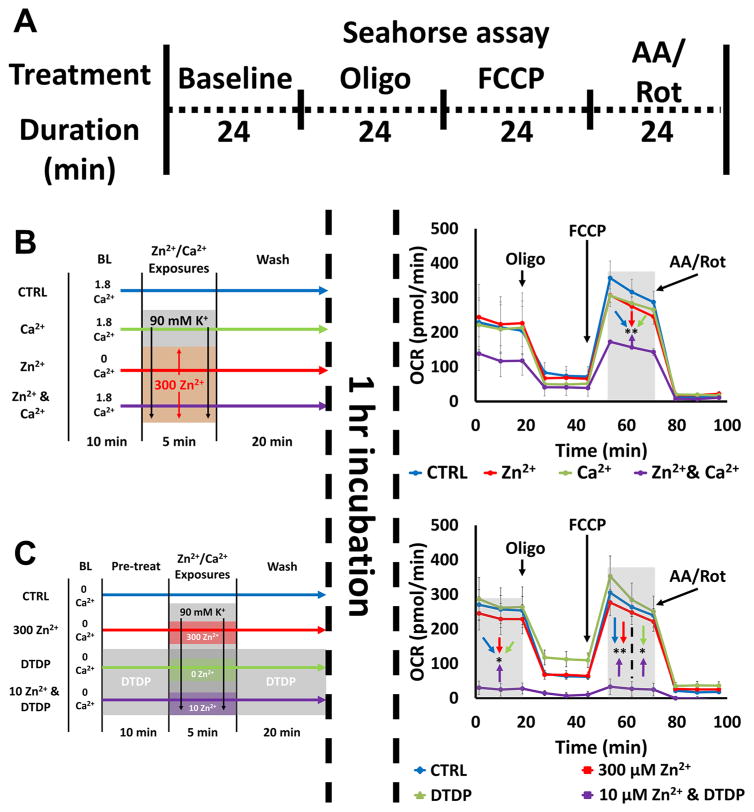
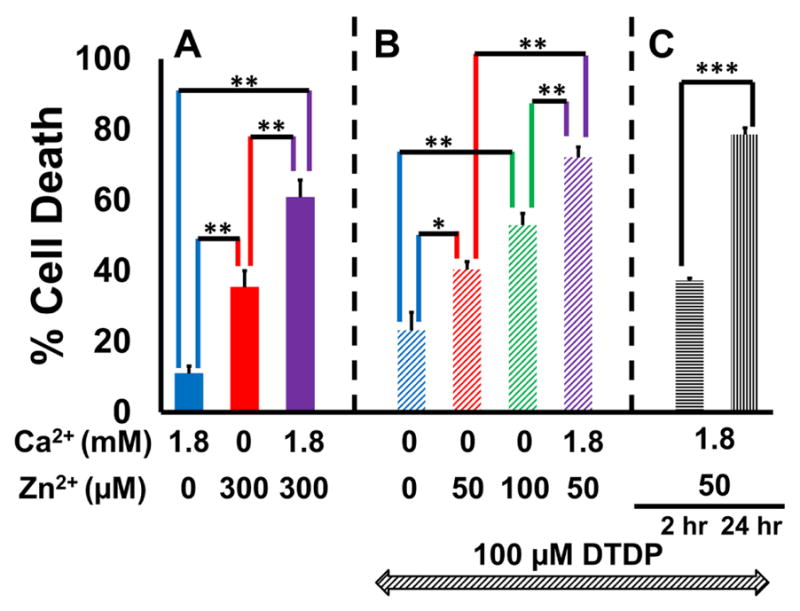
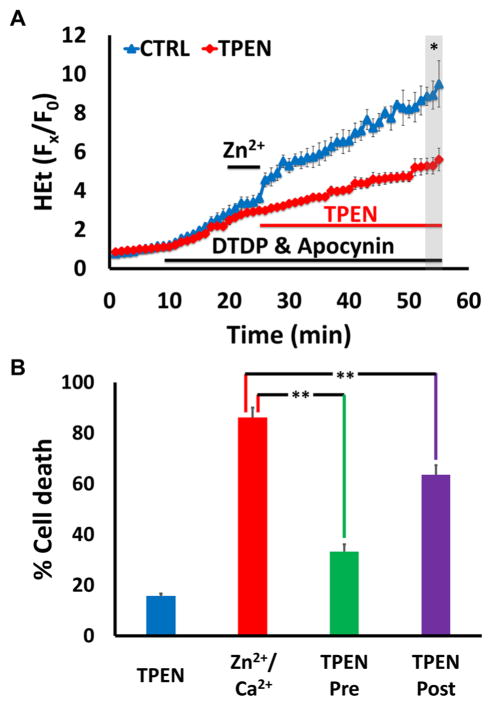
Excitotoxic Zn2+ and Ca2+ accumulation contributes to neuronal injury after ischemia or prolonged seizures. Synaptically released Zn2+ can enter postsynaptic neurons via routes including voltage sensitive Ca2+ channels (VSCC), and, more rapidly, through Ca2+ permeable AMPA channels. There are also intracellular Zn2+ binding proteins which can either buffer neuronal Zn2+ influx or release bound Zn2+ into the cytosol during pathologic conditions. Studies in culture highlight mitochondria as possible targets of Zn2+; cytosolic Zn2+ can enter mitochondria and induce effects including loss of mitochondrial membrane potential (ΔΨm), mitochondrial swelling, and reactive oxygen species (ROS) generation. While brief (5 min) neuronal depolarization (to activate VSCC) in the presence of 300 μM Zn2+ causes substantial delayed neurodegeneration, it only mildly impacts acute mitochondrial function, raising questions as to contributions of Zn2+-induced mitochondrial dysfunction to neuronal injury. Using brief high (90 mM) K+/Zn2+ exposures to mimic neuronal depolarization and extracellular Zn2+ accumulation as may accompany ischemia in vivo, we examined effects of disrupted cytosolic Zn2+ buffering and/or the presence of Ca2+, and made several observations: 1. Mild disruption of cytosolic Zn2+ buffering-while having little effects alone-markedly enhanced mitochondrial Zn2+ accumulation and dysfunction (including loss of ∆Ψm, ROS generation, swelling and respiratory inhibition) caused by relatively low (10-50 μM) Zn2+ with high K+. 2. The presence of Ca2+ during the Zn2+ exposure decreased cytosolic and mitochondrial Zn2+ accumulation, but markedly exacerbated the consequent dysfunction. 3. Paralleling effects on mitochondria, disruption of buffering and presence of Ca2+ enhanced Zn2+-induced neurodegeneration. 4. Zn2+ chelation after the high K+/Zn2+ exposure attenuated both ROS production and neurodegeneration, supporting the potential utility of delayed interventions. Taken together, these data lend credence to the idea that in pathologic states that impair cytosolic Zn2+ buffering, slow uptake of Zn2+ along with Ca2+ into neurons via VSCC can disrupt the mitochondria and induce neurodegeneration.
Keywords: Ca(2+) channel; Calcium; Excitotoxicity; Ischemia; Metallothionein; Mitochondria; Neuronal cultures; Reactive oxygen species; VSCC; Zinc.
Copyright © 2018 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Zn2+ entry through the mitochondrial calcium uniporter is a critical contributor to mitochondrial dysfunction and neurodegeneration.Exp Neurol. 2020 Mar;325:113161. doi: 10.1016/j.expneurol.2019.113161. Epub 2019 Dec 24. Exp Neurol. 2020. PMID: 31881218 Free PMC article.
-
Mitochondrial sequestration and Ca(2+)-dependent release of cytosolic Zn(2+) loads in cortical neurons.Neurobiol Dis. 2002 Jul;10(2):100-8. doi: 10.1006/nbdi.2002.0493. Neurobiol Dis. 2002. PMID: 12127148
-
Mechanisms of rapid reactive oxygen species generation in response to cytosolic Ca2+ or Zn2+ loads in cortical neurons.PLoS One. 2013 Dec 10;8(12):e83347. doi: 10.1371/journal.pone.0083347. eCollection 2013. PLoS One. 2013. PMID: 24340096 Free PMC article.
-
Rethinking the excitotoxic ionic milieu: the emerging role of Zn(2+) in ischemic neuronal injury.Curr Mol Med. 2004 Mar;4(2):87-111. doi: 10.2174/1566524043479211. Curr Mol Med. 2004. PMID: 15032707 Review.
-
Glutamate excitotoxicity and Ca2+-regulation of respiration: Role of the Ca2+ activated mitochondrial transporters (CaMCs).Biochim Biophys Acta. 2016 Aug;1857(8):1158-1166. doi: 10.1016/j.bbabio.2016.04.003. Epub 2016 Apr 7. Biochim Biophys Acta. 2016. PMID: 27060251 Review.
Cited by
-
The Multifaceted Roles of Zinc in Neuronal Mitochondrial Dysfunction.Biomedicines. 2021 Apr 29;9(5):489. doi: 10.3390/biomedicines9050489. Biomedicines. 2021. PMID: 33946782 Free PMC article. Review.
-
Mitochondrial Zn2+ Accumulation: A Potential Trigger of Hippocampal Ischemic Injury.Neuroscientist. 2019 Apr;25(2):126-138. doi: 10.1177/1073858418772548. Epub 2018 May 10. Neuroscientist. 2019. PMID: 29742958 Free PMC article.
-
A Neurotoxic Ménage-à-trois: Glutamate, Calcium, and Zinc in the Excitotoxic Cascade.Front Mol Neurosci. 2020 Nov 26;13:600089. doi: 10.3389/fnmol.2020.600089. eCollection 2020. Front Mol Neurosci. 2020. PMID: 33324162 Free PMC article. Review.
-
Zn2+ entry through the mitochondrial calcium uniporter is a critical contributor to mitochondrial dysfunction and neurodegeneration.Exp Neurol. 2020 Mar;325:113161. doi: 10.1016/j.expneurol.2019.113161. Epub 2019 Dec 24. Exp Neurol. 2020. PMID: 31881218 Free PMC article.
-
Protective effects of voltage-gated calcium channel antagonists against zinc toxicity in SN56 neuroblastoma cholinergic cells.PLoS One. 2018 Dec 20;13(12):e0209363. doi: 10.1371/journal.pone.0209363. eCollection 2018. PLoS One. 2018. PMID: 30571745 Free PMC article.
References
-
- Aizenman E, Stout AK, Hartnett KA, Dineley KE, McLaughlin B, Reynolds IJ. Induction of neuronal apoptosis by thiol oxidation: putative role of intracellular zinc release. J Neurochem. 2000;75:1878–1888. - PubMed
-
- Assaf SY, Chung SH. Release of endogenous Zn2+ from brain tissue during activity. Nature. 1984;308:734–736. - PubMed
-
- Bonanni L, Chachar M, Jover-Mengual T, Li H, Jones A, Yokota H, Ofengeim D, Flannery RJ, Miyawaki T, Cho CH, Polster BM, Pypaert M, Hardwick JM, Sensi SL, Zukin RS, Jonas EA. Zinc-dependent multi-conductance channel activity in mitochondria isolated from ischemic brain. J Neurosci. 2006;26:6851–6862. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
